Abstract
5-Fluorouracil (5-FU) is the leading chemotherapeutic drug used to treat hepatocellular carcinoma, one of the major cancer diseases after atherosclerosis. Because of chemo-resistance, the success rate of treatment declines with time due to continuous drug exposure. Though autophagy induction is majorly responsible for acquired resistance, the exact role of this evolutionary conserved mechanism is unknown in cancer cell survival and suppression. The usual practice involves the combinatorial use of chemotherapeutic drugs with autophagy inhibitors like Chloroquine and Bafilomycin A, while neglecting the side effects caused by autophagy impairment in healthy cells. Starvation is a well-known physiological inducer of autophagy. In this study, by caloric modulation, we tried to circumvent the resistance imposed by prolonged drug exposure and investigated the effect of 5-FU in nutrient-sufficient and deficient conditions. Our findings show a substantial correlation between autophagy and increased cancer cell death in the presence of 5-FU, with negligible effects on normal cells. Experimental data revealed that nutritional deprivation augmented cell death in the presence of 5-FU through mitochondrial membrane damage and excessive reactive oxygen species (ROS) production, initiating apoptosis. Lipidation study also unveiled that under such combinatorial treatment cellular metabolism shifts from glucose to lipid biosynthesis. Overall, our experimental findings suggest that nutritional deprivation in combination with chemotherapeutic medication can be a new effective strategy to control hepatocellular carcinoma.
Introduction
Hepatocellular carcinoma (HCC) is one of the most devastating reason involves behind cancer-related fatalities1. According to the World Health Organization, by 2025, > 1 million individuals will be affected by HCC2. In this scenario, the ultimate concern involves the late diagnosis of HCC3, which limits the surgical option to fewer than 20% of patients4. Therefore, systemic chemotherapy emerges as a major therapeutic approach to treat liver cancer5. In this context, the use of 5-Fluorouracil (5-FU) remained the first choice of defense against advanced HCC. But, its application is limited due to increasing chemo-resistance6,7. Recent studies suggested that the acquired resistance is somehow related to autophagy8,9,10,11 and can be reversed by applying 5-FU along with other chemotherapeutic drugs like sorafenib and forbesione12,13. However, it has not yet been reported how this combinatorial strategy affects normal cells. Autophagy is a catabolic process that eliminates non-essential or burned-out organelles, proteins, and foreign entities inside the cell to generate pool of bio-molecules to produce ATP for cell survival14. The upregulation of basal autophagy occurs under stressful conditions like nutrient deprivation, ER stress, and pathogen infection. During chemotherapy, autophagy has been considered a double-edged sword for providing both chemo-resistance and chemo toxicity15. Sometimes it acts as an adaptation process that confers resistance against chemotherapy10. On the other hand, increasing basal autophagy to chemo-sensitize cell is now emerged as another unconventional and useful tactic for combating apoptosis resistance in some cancers9. In those cases, the resultant autophagic cell death is distinctive from other programmed cell death9,16. According to earlier researchers, combinatorial treatment of autophagy inhibitors with anticancer drug provides promising result by slowing cancer cell proliferation17. However, no studies have been reported to determine how such treatment affects normal cells. In this direction, the usual practice includes the application of inhibitors like chloroquine, 3-methyladenine and hydroxychloroquine etc17. , ignoring the fate of normal cells. The prolonged applications of non-selective inhibitors are quite detrimental to normal cell growth, as these inhibitors eventually hinder basal autophagy. In fact, autophagic dysfunction has been coupled with various neurodegenerative disorders like parkinson’s disease, and lysosomal storage disorders18. It is exceedingly unlikely that pharmacological inhibitors would function in a tumor-selective manner; therefore, prolonged and persistent repression of autophagy could result in unwanted and possibly life-threatening side effects18. As a result, this could cause permanent harm as a consequence of chemotherapeutic side-effects.
Considering its dual impact, autophagy induction coupled with chemotherapeutic drug now emerges as a breakthrough strategy to overcome drug resistance19,20. Preclinical studies propose that fasting prior to chemotherapy may be an effective strategy to protect patients against the unpleasant effects of chemo-toxicity21,22 and it may also sensitize cancer cells towards chemotherapy. In turn, such fasting cues would prompt normal cells to refocus their energy on cellular maintenance and repair activities, rather than growth and proliferation23. Fasting also encourages autophagy-reliant epitope processing, which strengthens immune-surveillance23. Emerging preclinical evidence confirms that short-term fasting (STF) enhances the potency of a wide number of chemotherapeutic agents24. Under such conditions, healthy cells are more resilient to the stress imposed by STF, while tumour cells become more susceptible to toxins, presumably due to the shortage of nutrients to meet cancer cells’ requirements in the context of high proliferation rates24,25. In humans, STF can be considered as a practical strategy to improve the efficacy and admissibility of chemotherapy. It appears safe if used in conjunction with chemotherapy in humans, and may also lessen numerous side effects in healthy cells25. To thoroughly establish clinical efficacy and safety, more study in this direction is necessary. Considering all, here we sought to induce autophagy by complete nutrient deprivation and investigated its effect in combination with the chemotherapeutic drug 5-FU as a possible therapeutic strategy to treat hepatocellular carcinoma.
Results
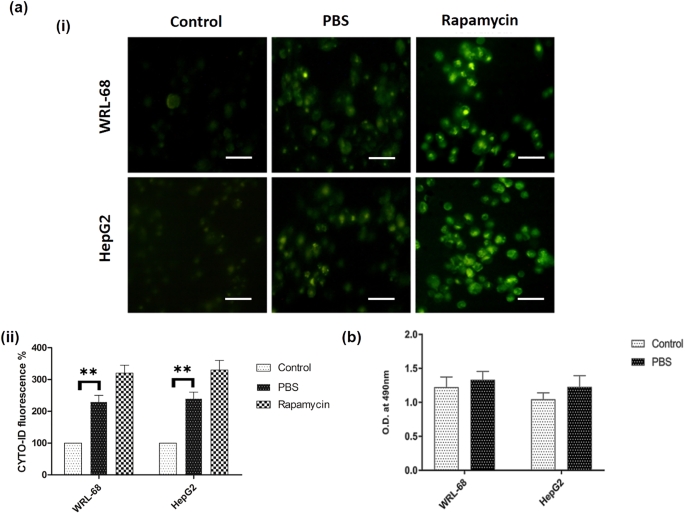
Induction and assessment of autophagy in WRL-68 and HepG2 cell lines
Autophagy-mediated cell death mechanism contributes to the efficacy of anticancer drugs9,14. With the potential of autophagy to either induce cell death or promote cell survival10,26, a paradoxical role of autophagy following anticancer treatment was studied at different time intervals (0–3 h). The autophagy was induced by complete nutrient starvation and confirmed by CYTO-ID®. In Fig. 1(a), bright green fluorescence was recorded after 3 h of treatment in nutrient starved cells (PBS treated). However, during the initial 2 h there was no such positive response related to autophagy was observed (data not shown). After 3 h of starvation, an observable fluorescent intensity was clearly noticed in both cell lines. Here, cells treated with rapamycin (autophagy inducer) were considered as positive control. Therefore, successful autophagy induction through complete nutrient starvation was achieved in all cell lines. The role of autophagy in cancer cell death and survival remains controversial to date27. Literature suggests that autophagy induction can be a breakthrough strategy to augment cytotoxic drug activity9,27,28 in various malignancies. Thus, to investigate the impact of autophagy, we evaluated how nutritional deficiency modulates cell viability. In Fig. 1(b), LDH assay was performed to notice the impact of PBS on cell viability and we found no significant effect up to 3 h. Furthermore, we determined intracellular stress response enzymes’ level to ascertain cellular homeostasis.

Autophagy induction via complete nutrient starvation: (a) Autophagy induced in normal (WRL-68) and cancer cell (HepG2) lines. (i) Formation of autophagosomes were detected by CYTO-ID® autophagy detection kit in presence of complete media (control), PBS and rapamycin (inducer of autophagy) in both cell lines, (ii) Quantitative change in fluorescence intensity relative to control. (b) LDH assay was performed in complete media (as control) and PBS and the results are represented as ± SD of at least three independent experiments. p < 0.05 was considered as significant; the standard deviations of the data have been shown in the form of error bars. Scale bar = 30 µM.
Determination of intracellular ROS, GST, NO, NO2 and NO3 production
Metabolic by-products like ROS are extremely unstable and involved in maintaining cellular homeostasis through cell-signalling29. The detection of ROS levels largely relies on detecting the end products by H2DCFDA staining30. As compared to rapamycin and H2O2, the fluorescence intensity was less in PBS (starvation) treated cells. Negligible fluorescence was detected in cells having complete media (control) Fig. 2(a). In both cell lines, no significant variation indicates less homeostatic changes due to time bound PBS exposure. As nutrient deficiency can lead to a decrease in glutathione levels in cells31, we examined its expression after 3 h of treatment and compared the following expression with cells under normal growth-supporting media. Results from Fig. 2(b), indicates a significant change in GST level in both cell lines. In the absence of nutrient, there is a slightly elevated expression of GST was quantitatively determined particularly in HepG2. Interestingly, a decreased expression of GST was recorded in the WRL-68 cell line upon starvation. Thus, the differential stress response of normal and cancer cell lines towards nutrient starvation was clearly noted. Literature also suggests that under autophagic condition, the expression level of GST is closely associated with signalling molecules like NO32. Moreover, NO is also considered as an inducer of autophagy by causing the activation of AMPK32. Therefore, the total nitric oxide level was quantified based upon the reduction of NO3‾ to NO2‾ by reducing agent at 37 oC. Endogenous nitrite and nitrate were collectively converted by Griess Reagent to a pink-coloured azo compound that was measured spectrophotometrically at 580 nm. From the curated data [Fig. 2(c), 2(d), and (e)] it was found that in both the nutrient-starved cells, NO levels were elevated as compared to their respective control.

Cellular ROS, GST, and nitric oxide determination in autophagic cells: (a) ROS production was determined by 2’-7’-Dichlorodihydrofluoroscein diacetate (H2DCFDA) dye (2 µg/ml) in presence of complete media control), PBS, H2O2, and rapamycin. (i) All the cells were observed using fluorescence microscope (Magnus MLXi, India). Images included here are representative of three independent experiments on both cell lines. (ii) Quantitative change in fluorescence intensity relative to control. (b) (c) (d) & (e) Stress response enzyme assay was performed by quantitative measurement of glutathione’s transferase, nitric oxide, nitrite and nitrate activity, respectively. Values are represented as ± SD of at least three independent experiments. p < 0.05 was considered as significant; the standard deviations of the data have been shown in the form of error bars. Scale bar = 30 µM.
Expression profile of autophagy-targeted genes (ATGs) in WRL-68 and HepG2 cell lines
For the specific detection of autophagy-related genes we have performed indirect ELISA assay33. It was observed that the expression level of β-actin (housekeeping gene) was not affected in both cell lines [Fig. 3(a) and (b)]. Whereas, significant Beclin1 expression was seen in starved condition, reflecting a successful autophagic initiation process. There was an elevated expression of Atg3 in autophagic cells was also recorded. It indicates that E2 enzyme (Atg3) is needed for LC3 lipidation which is involved in forming Atg16L complex, to perform E3-like role in the LC3 lipidation reaction14. From the depicted graph in Fig. 3(a) and (b), it was evident that in nutrient-deficient condition, autophagy-related proteins like – Atg7, Atg5, and LC3B were in higher concentration compared to non-autophagic cells. These over-expressed biomarkers are particularly related to macro-autophagy14 and the different expression patterns indicated their different susceptibility towards autophagy induction.

ATGs (Autophagic Targeted Genes) expression profile in autophagic WRL-68 and HepG2 cell lines: (a) and (b) Autophagy progression was determined in control (cells without treatment), and PBS (nutrient deprived for 3 h) by antibody based indirect ELISA method. The cytosolic protein was isolated from each treated condition and was subjected to indirect ELISA for quantifying autophagy related biomarkers. Quantification was achieved by recording absorbance at 450 nm using SPECTROStar Nano plate reader (BMG Labteck, Germany). All data are representative of three independent experiments and are expressed as mean ± SD. p < 0.05 was considered as significant; the standard deviations of the data have been shown in the form of error bars.
Differential response of 5-FU under autophagic environment
The role of autophagy in cancer cell death and survival remains controversial to date27. Literature suggests that autophagy induction can be a breakthrough strategy to augment cytotoxic drug activity9,19,34. Thus, to investigate the impact of autophagic condition, we evaluated how nutritional deficiency modulates cell viability under 5-FU treatment. At first, cells were treated with increasing concentrations of the drug (50–500 µM) for 24 h [Fig. 4(a)] and the IC50 doses were determined accordingly (for WRL-68 250 µM and for HepG2 cells 300 µM ). Next, a trypan blue exclusion assay was performed after 3 h for the comparative analysis of viable and non-viable cells in PBS alone and in combination with the chemotherapeutic drug. Here, we have found [Fig. 4(b)] that nutrient starvation did not cause an immense decrease in cell viability for a particular period of time. The surviving ability of both cells was not significantly affected up to 3 h. However, when the cells were treated with 5-FU, the drug efficacy was more pronounced in cancer cells under starvation as compared to the normal cells [Fig. 4(b)]. The morphological changes were also captured under each condition [Fig. 4(c)]. In both cell lines, a major morphological difference was observed in cells treated with PBS + 5-FU, indicating a strong cellular response against the treatment. Furthermore, we investigated the presence of AVOs to determine whether 5-FU is only responsible for inducing significant autophagy-mediated toxicity or not. Here, Fig. 4(d) clearly indicated the presence of AVOs not only in PBS and rapamycin (5 µM) treated cells but also in cells treated with 5-FU under normal growth supporting media. However, excessive AVO formation was clearly recorded in cells under the combinatorial effect of PBS + 5-FU.

The comparative cell viability study in starved autophagic cells in presence of 5-FU: (a) The percentage of cell viability after 5- Fluorouracil treatment in WRL-68 and HepG2 cell by MTT assay after 24 h. IC50 dose of 250 µM and 300 µM were determined for WRL-68 and HepG2, respectively. (b) Trypan blue exclusion assay was performed for the accurate quantification of viable and non-viable cells in all treated conditions – control, PBS only (without nutrient), control + 5-FU and PBS + 5-FU (starvation induced autophagic condition + 5-Fluorouracil). All the values plotted are the averages of three different experiments; the standard deviations of the data have been shown in the form of error bars. (c) Morphological changes of HepG2 cell line and WRL-68 were observed under phase contrast microscope at initial stage (0 h) and after successful autophagy induction (3 h). All the experiments were performed in triplicate. Scale bar = 80 µM. (d) Autophagolysosomes were detected by the formation of acidic vacuole organelles under following conditions– complete media(control), nutrient deficient condition (PBS), 5-FU treated cells in presence/absence of nutrient (control + 5-FU and PBS + 5-FU) and Rapamycin treated cells (as positive control). All the experiments were done in triplicate and images represented are representative of three individual treatments. Scale bar = 30 µM.
Impact of 5-FU on cellular redox balance and lipidation metabolism under autophagic condition
As cell death is generally associated with reactive oxygen species formation35, we determined ROS by staining cells with DHE dye after PBS + 5-FU treatment. Figure 5(a) demonstrated a significant ROS production in all cell lines exposed to 5-FU combined with PBS (PBS + 5-FU). Interestingly, a major contribution to ROS production comes from PBS + 5-FU treated HepG2 cells when compared with WRL-68. According to reports, increased formation of ROS, ER stress, and lipotoxicity are all caused by hepatocytic lipid degradation36. Under starvation, autophagy (particularly macrolipophagy) has been found to regulate intracellular lipid storage through controlled degradation of lipid droplets and release of fatty acids into the cytosol37,38. Therefore, we investigated the lipidation status of both cell lines under each mentioned condition [Fig. 5(b)]. From the data, we recorded excessive lipidation in both starved cell lines. Figure 5(b) suggested that under the combinatorial effect of PBS + 5-FU, intracellular lipid degradation is much higher in the HepG2 cell line. A similar finding was also noted in the normal hepatic WRL-68 but to a limited extent. Our quantitative measurement also validates the microscopic observation [Fig. 5(c)].

Detection of reactive oxygen species formation using Dihydroethidium (DHE) dye. (a) Superoxide formation was detected by staining the cells with DHE (2 µg/ml) in presence and absence of nutrient along with 5-Fluorouracil. (i) WRL-68 and HepG2 cells were visualized under fluorescence microscope (Magnus MLXi, India). (ii) Quantitative change in fluorescence intensity relative to control. Scale bar = 50 µM. (b) Qualitative measurement of cellular lipidation status under each mentioned condition –complete media(control), PBS (nutrient deficient condition), 5-Fluorouracil under nutrient sufficient (control + 5-FU) and nutrient deficient condition (PBS + 5-FU). All stained cells were observed under a fluorescence microscope (Magnus MLXi, India). Scale bar = 30 µM. (c) Differential lipidation pattern was observed in – Control (without treatment), PBS (Nutrient deficient condition), 5-Fluorouracil under nutrient sufficient (control + 5-FU) and nutrient deficient condition (PBS + 5-FU). Extraction of the dye was done with 100% isopropanol and absorbance was recorded at 492 nm by SPECTROstar nano plate reader (BMG Labteck, Germany). 100% isopropanol was used as background control to subtract the background signal. All the values plotted are the averages of three different experiments; the standard deviations of the data have been shown in the form of error bars.
Strategically autophagic cells in presence of 5-FU-induced apoptosis
We performed annexinV-FITC/PI staining for the accurate measurement of cell death and found that [Fig. 6(a)] the combination of PBS and 5-FU forms very few early apoptotic bodies in WRL-68. Whereas, an abundance of late apoptotic cells stained with propidium iodide confirmed the occurrence of apoptosis in HepG2 cancer cells. Since depolarization of mitochondrial homeostasis can result in cell death29, we examined the mitochondrial membrane damage of TMRE stained cells in different conditions. Our analysis reported in Fig. 6(b) suggests that in WRL-68, PBS alone did not cause massive mitochondrial membrane damage when compared to control. Depolarization or damaged mitochondria was observed in the presence of 5-FU. Both 5-FU andPBS + 5-FU treated cells indicated a possible sensitization to chemotherapeutic treatment. Notably, the observable damage found in association of PBS with 5-FU treatment is quite higher in HepG2 than WRL-68.

Detection of mitochondrial membrane damage through TMRE Assay. (a) Assessment of cell death was separately recorded in WRL-68 (normal liver cell line) and HepG2 (Hepatocarcinoma cell line) by Annexin V-FITC/PI conjugate assay- WRL-68 and HepG2 cells were treated with 5-FU drugs in complete media (control), PBS for 3 h and stained according to manufacturer’s protocol. (i) Slides were visualized under a fluorescence microscope (Magnus MLXi, India) and all the figures are representative of three individual experiments. (ii) Quantitative change in fluorescence intensity relative to control. Scale bar = 100 µM. (b) WRL-68 and HepG2 cells were treated with 5-FU drug in complete media (control) and with PBS. Cells were stained with 50 nM of tetramethylrhodamine ethyl ester (TMRE) dye. (i) Cells were observed using a fluorescence microscope (Magnus MLXi, India).and all the images included here are representative of three independent experiments on both cell lines. (ii) Quantitative change in fluorescence intensity relative to control. Scale bar = 30 µM. (c) Acridine Orange/Ethidium Bromide (2 µg/ml) staining was performed to detect apoptosis in cells under the combinatorial effect of PBS + 5-FU (5-Fluorouracil under nutrient deprived condition). (i) Early and late apoptotic cells were detected by staining with acridine orange and ethidium bromide dye using a fluorescence microscope (Magnus MLXi, India). Presence of apoptotic cells were compared with that of untreated control (with growth media) by three independent experiments. (ii) Quantitative changes in red and green fluorescence intensity. Scale bar = 30 µM.
From the acridine orange/EtBr staining we noticed significant green fluorescence in the control panel [Fig. 6(c)]. However, ethidium bromide uptake in presence of 5-FU was clearly visible after 3 h [Fig. 3(a)]. There was a higher number of late apoptotic HepG2 cells found under the combinatorial treatment (PBS + 5-FU). The bright red fluorescence suggests that apoptosis initiation leads to the ethidium bromide uptake, particularly in HepG2 cells. Interestingly, very few apoptotic cells emitting red fluorescence were visualized in WRL-68. Also, the higher green fluorescence emission from viable WRL-68 ensures the uptake of acridine orange only. On the contrary, the abundance of non-viable HepG2 cells was clearly distinguished by its bright red fluorescence.
Discussion
The survival rate of HCC is compromised due to delayed and poor prognosis1. Moreover, the use of 5-FU against HCC is limited by its inherent toxicity to normal cells and the development of chemo-resistance5,6. Although the mechanism of chemo-resistance is being widely investigated to develop novel target-based therapies, but more effective therapeutic strategies must be explored to combat the flexibility of acquired drug resistance. According to reports, as autophagy is responsible for this acquired resistance14, hence several inhibitors are used in addition to chemotherapeutic drugs, but the detrimental consequences of autophagy impairment in healthy cells are overlooked32. Since normal cells’ autophagic machinery can be imbalanced by extended exposure to autophagic inhibitors this type of combinatorial treatment should be avoided. However, reports in this direction are somehow limited. From ancient times, fasting or starvation has attracted the most interest for its potential to improve health and lengthen healthy lifespans in a variety of animals, including primates33. Therefore, modulating this usual practice we developed a new efficient and selective strategy to abolish the previously discussed shortcomings. Here, we established autophagy by complete nutrient starvation in both WRL-68 and HepG2 cells to detect the differential response of normal and cancer cells under autophagic environment. In this study, preliminary evidence of autophagy progression was achieved by detecting autophagosomes through CYTO-ID® [Fig. 1(a)] staining and measuring the quantitative expression of each autophagy associated biomarker through indirect ELISA [Fig. 3(a) and 3(b)]. A robust cellular response to stress imposed by total nutrient withdrawal is indicated by the elevated expression of autophagy-associated antibodies. Therefore, by using this biological autophagy inducer (starvation) we successfully established autophagic cell lines to mitigate chemo-resistance by prolonged drug exposure39. Our data also clearly demonstrates a differential expression pattern which might be due to the varied susceptibilities of these cell lines in response to autophagy28. Notably, as Beclin1 is considered an anti-apoptotic gene36, therefore its augmentation also indicates that nutrient deprivation alone does not cause cell death. Under such conditions, morphological alterations were quite apparent without any loss in cellular viability [Fig. 1(b), 4(b) and 4(c)].
Excessive AVO formation under the combinatorial treatment of PBS + 5-FU also suggests that chemotherapeutic drug in presence of culture media alone was not capable of boosting basal autophagy to the similar extent that 5-FU did in PBS-treated cells [Fig. 4(d)], specifically in HepG2. This observation highlights an intriguing finding that suggests 5-FU acts differently in normal and cancer cells during starvation. To assess the degree of intracellular stress response, quantification of GST [Fig. 2(b)] and nitric oxide were alsodone, [Fig. 2(c), 2(d) and 2(e)]. The obtained result revealed that the reduced GST activity particularly in the nutrient starved normal cells (WRL-68) might be due to the fact that fasting has halted several metabolic processes that would otherwise produce toxic end products37. During starvation, cellular growth and proliferation stop, and the primary goal of cells shift towards survival. Thus, cells tend to turn on the activity of only those components that are indispensable for survival. When there is an inadequate supply of nutrients, this selective activation benefits the cell in preserving resources and energy. However, in cancer cells, the GST activity was slightly elevated which is consistent with the findings of higher reactive oxygen species formation [Fig. 2(a)]. This observation suggests that in order to counteract ROS-mediated damage, cells were producing maximum anti-oxidants (GST) for their survival40. Literature suggested that in several tumors, increased GST activity contributes to treatment resistant and involves in the metabolism of endogenous lipid mediators31. GSTs also influence diverse signalling pathways that modulate cell survival during stress condition. Additionally, they are also involved in the cascade of cell proliferation31. So, the elevated level of GST in HepG2 cells might be acting as a signalling molecule in overcoming the stress response and counteract excessive ROS production. Nitric oxide, another biological signalling molecule associated with autophagic modulation has also been estimated in higher concentration, particularly in starved cells [Fig. 2(c), 2(d) and 2(e)]. NO has been demonstrated to be an inducer of autophagy by causing activation of AMPK38. It serves as a regulator of autophagy through ATM-AMPK-TSC2–mediated suppression of mTORC138,41. Therefore, it is evident from the increased nitric oxide level that the nutrient-deficient cells were actively attempting to survive by activating the nutrient sensor protein AMPK and suppressing mTORC1. Subsequently, this activation-suppression cascade initiates the autophagic process. Following the effective autophagy induction, we investigated whether autophagic induction by starvation also manifests chemotherapeutic resistance. Surprisingly, we found that starvation-induced autophagy increases the effectiveness of 5-FU in all cell lines, but especially in hepatic cancer cells (HepG2) that were previously less susceptible to 5-FU alone [Fig. 4(b) and 4(c)]. Although a slight decrease in normal cell (WRL-68) reproducibility was also recorded under the combinatorial effect (PBS + 5-FU). This observation suggests that the approach will be well tolerated by normal cells. Moreover, we assessed the autophagosome accumulation in cells treated with 5-FU alone and compared it with other treatment conditions. This evaluation aimed to ascertain the extent to which 5-FU alone induce an autophagic response. This comparative analysis also demonstrated the differential response of cells towards chemotherapeutic drug upon autophagy induction. Data curated from Fig. 4(d) suggests that 5-FU is itself capable of generating acidic vacuole organelles (AVOs) similar as rapamycin (positive inducer of autophagy), which corroborates the problem that arise during 5-FU resistance chemotherapy10. But, cells treated with 5-FU under starvation condition suggest that the increasing cytotoxicity observed in HepG2 is due to the upregulated autophagy induced by PBS which boosts chemotherapeutic drug toxicity. Consequently, the aforementioned findings proved that the cancer cells (HepG2) are more susceptible to such treatment when compared to normal cells (WRL-68). However, more studies are required to firmly establish this strategy. According to previous reports, autophagy stimulation boosted the effectiveness of chemotherapeutic drugs in a variety of tumour cells19. However, the precise mechanism has not explored yet due to the varying susceptibility of cells toward chemotherapeutic treatment42,43. Here, by further investigating the cell death mechanism, we found that excessive reactive oxygen species accumulation [Fig. 5(a)] causes mitochondrial membrane damage [Fig. 6(b)] in HepG2. The damaged mitochondria are unable to sequester the positively-charged, cell-permeant red coloured dye TMRE and got affected more prominently under the treatment condition (PBS + 5-FU). Moreover, such imbalance in mitochondrial homeostasis resulting in cell death is often accompanied by an increase in ROS generation during the onset of apoptosis29. A higher number of late apoptotic bodies found in PBS + 5-FU treated cells also confirmed cellular apoptosis, particularly in HepG2 cells [Fig. 6(c)]. This differential mortality pattern reveals a time and dose-dependent relationship between drug cytotoxicity and autophagy during tumour cell suppression. Therefore, our finding indicates that excessive ROS production as a result of depolarized mitochondrial status drives HepG2 cells toward apoptosis. Under this circumstance, we investigated whether cellular lipidation, rather than carbohydrate metabolism was playing any significant role or not. We performed oil red o uptake assay on these two hepatic cell lines to identify any alteration related to drug detoxification and lipidation [Fig. 5(b)]. Reports suggested that inflammatory, proliferative and apoptotic signals inside this detoxifying organ are closely interlinked. These interconnected pathways play a crucial role in maintaining hepatic lipid and glucose metabolism44. As hepatic lipid homeostasis is modulated by autophagy, therefore under fasting, the lysosomal route recycles expendable cellular constituents into a vital energy source and provides chemoresistance45 to normal hepatic cells (WRL-68).
Additionally, recent research on animals shows that autophagy induction is a vital step in the breakdown of lipid droplets and hepatic lipolysis45. Nutrient deficiency augments macro-autophagy which causes the lysosome and lipid droplets to fuse into autophagolysosome which are then subsequently broken down. The fatty acids are then liberated and utilized for metabolic reactions to maintain normal cell viability under stress condition43. Here, the hepatic cancer (HepG2) cells were also trying to mitigate the stress condition via lipid degradation but came out unsuccessful. The unavailability of glucose source forced them to utilize its lipid storage and due to loss of flexibility in the context of high proliferation rates, cancer cells were unable to do so22,23. Therefore, all our data demonstrates that excessive stress imposed via nutrient starvation along with chemotherapeutic drug (5-FU) not only increases cytotoxicity but also exerts cell transition from glucose to lipid metabolism. Here, we used in vitro model for understanding the perplexity of starvation-induced autophagy to increase chemosensitivity. But, as the system does not accurately mimic the complexity of in vivo system, treatment outcome may vary in terms of immune response, tissue architechture and systemic metabolism. Though this study provides a strong foundation in evaluating the role of autophagy under chemotherapy, research endeavours ought to integrate animal models to ensure the clinical safety and efficacy of this intervention within a more comprehensive biological system.
Conclusions
Our elaborate experimental findings demonstrate that complete nutritional deprivation (using PBS) with 5-FU was quite effective in enhancing cancer cell death, with a bearable influence on normal hepatic cell. We also found that autophagy is particularly involved in this increased drug-mediated cytotoxicity followed by excessive ROS accumulation resulting from damaged mitochondria inducing apoptosis. Lipolysis also plays a significant role, particularly under the combinatorial treatment. However, further study in this direction is necessary to unveil other cellular mechanisms that may influence cancer cell death by nutritional shortage and chemotherapeutic treatment. Finally, all the above findings indicate that caloric intake modulation in combination with chemotherapeutic medication will be a new effective, and breakthrough strategy to control hepatocellular carcinoma.
Materials and methods
Cell lines and treatment
WRL-68 and HepG2 cells were procured from National Centre for Cell Science (NCCS); Pune (India) and were grown in DMEM (Dulbecco’s Modified Eagles Medium; Hi-media Laboratory Pvt. Ltd., India) media with 10% FCS (Fetal Calf Serum; Hi-media Laboratory Pvt. Ltd., India) at 37 °C in humidified atmosphere containing 5% CO2. The growth medium was supplemented with 10 µg/ml of penicillin G and 10 µg/ml of streptomycin (Sigma-Aldrich, USA). Splitting/passage was performed by trypsinizing cells with 1X Trypsin-EDTA (Hi-media Laboratory Pvt. Ltd., India) when cells were at appropriate confluency (80-90%). Complete nutrient starvation was induced by replacing culture media with PBS (Phosphate buffered saline; Hi-media Laboratory Pvt. Ltd., India). 5-FU (CAS 51-21-8, F6627, Sigma-Aldrich, USA) was dissolved in dimethyl sulphoxide (DMSO) to 10 mM stock solution used for treatment. CYTO-ID® autophagy detection kit was purchased from Enzo Life Sciences, USA. Rapamycin (500 µΜ) was being provided in the kit (ENZ-51031-0050). GST (glutathione-s-transferase) assay kit (CS0410) and Nitric oxide (SKU23479-1KT-F) kit and Lactate Dehydrogenase Assay (LDH assay) kits (MAK066-1KT) were purchased from Sigma-Aldrich, USA. A lipid (Oil red O) staining kit was also purchased from Sigma-Aldrich, USA (MAK194). All cell line-based biological experiments were performed in triplicates.
Antibodies
The following antibodies were used – Beclin-1 rabbit mAb 3495 (Cell Signalling Technology, D4065), LC3A/BXP® Rabbit mAb #12,741 (Cell Signalling Technology, D3U4C), Atg5 rabbit mAb #12,994 (Cell Signalling Technology, D5F5U), Atg12 rabbit mAb #4180(Cell Signalling Technology, D88H11), Atg16L1 rabbit mAb #8089 (Cell Signalling Technology, D6D5), Atg7 rabbit mAb #8558 (Cell Signalling Technology, D12B11), Atg3 rabbit mAb #3415 (Cell Signalling Technology) and Anti-rabbit IgG, HRP-linked Antibody #7074. ß-actin was taken as a housekeeping gene (Santa Cruz Biotechnology, SC 47778).
Autophagosome detection by CYTO-ID® autophagy detection kit
To determine autophagosomes, a CYTO-ID® autophagy detection kit was used46. Briefly, cells were grown and treated with PBS for 3 h. Staining was done according to the manufacturer’s protocol with slight modification (ENZ-51031-0050) and observed under a fluorescence microscope (Magnus MLXi, India) with excitation at 480 nm and emission was photographed with Olympus digital camera. Rapamycin (5 µM) was used as a positive control. The fluorescence emission was recorded using three biological replicates.
LDH assay
HepG2 and WRL-68 cell lines were cultured at 37 °C with 5% CO2 for 24 h. Next day, culture media was replaced with PBS for 3 h and LDH assay was performed according to the manufacturers’ protocol47. In brief, lysis solution was added for 40 min and cell supernatant was collected from each treated and untreated samples. The collected supernatant was then allowed to react with the LDH reagent in a 1:1 concentration. After 30 min, a stop solution was applied and absorbance was recorded by a spectrophotometer (SPECTROstar nano, BMG Labtech, Germany) at a wavelength of 490 nm (main wavelength) and 600 nm (reference wavelength). The amount of LDH release was then compared with each control.
Detection of intracellular reactive oxygen species formation (ROS)
Formation of intracellular H2O2 was determined by staining the cells with DCFDA dye (2’-7’-Dichlorodihydrofluoroscein diacetate) for 1 h in the dark30. Before that, both the cell lines were exposed to nutrient-deficient condition at least for 3 h. H2O2 and rapamycin-treated cells were taken as positive inducer of ROS and autophagy, respectively. All slides were prepared carefully and observed under a fluorescence microscope (Magnus MLXi, India) at an excitation wavelength of 480 nm and emission was photographed with an Olympus digital camera.
Cellular homeostasis determination in nutrient sufficient and nutrient deficient condition
Stress response enzymes’ activity was determined by using GSTassay kit and Nitric oxide kit according to the manufacturer’s protocol with minor modifications48. Enzyme activity was calculated using the following formula:
GST activity {(µM/ml)/min} = ΔA340 X reaction volume X sample dilution factor/ extinction coefficient of GSH-DNB adduct X sample volume added to well (ml).
The concentration of Total Nitric Oxide and Nitrite was determined using the following equation of the standard curve:
Total nitric oxide (µM) = Corrected absorbance –Y intercept/Slope.
Nitrite (µM) = Corrected absorbance –Y intercept/ Slope.
Nitrate (µM) = Total Nitric Oxide – Nitrite.
Atg (autophagy related genes) expression profile detection by Indirect ELISA (enzyme-linked Immuno- Sorbent assay)
Proteins isolated from treated and untreated cells (both cell lines) using RIPA lysis buffer were estimated through Bradford’s protein quantification method. 100 ng of the freshly isolated protein was coated in an ELISA (NUNC) plate by using coating buffer (0.5 M carbonate-bicarbonate buffer, pH 9.5) and left overnight at 4 °C. Next day, the plate was washed to remove any unbound protein and allowed to dry. Thereafter, a blocking solution (1% Bovine Serum Albumin in PBS) was added and the incubation process was continued for a further 2 h at room temperature. Primary monoclonal antibody solution (1:1000 dilution for ß-actin antibody and 1:100 for Autophagy associated antibodies) was given after the removal of the blocking solution. Next day, HRP- conjugated secondary antibody (1:500 dilution) was used, to which OPD substrate solution (O-Phenylenediamine Dihydrochloride in citrate buffer) was added to record the absorbance (SPECTROstar nano, BMG Labtech, Germany) after 2 h at 450 nm wavelength49. All differential expressions reported had three biological replicates.
Cell viability determination by MTT assay
For determining the reproducibility of cells MTT assay was performed50. Approximately, 1 × 104/ml cells were added in a 96-well plate and incubated with different concentrations of 5-FU drug (50–500 µM) at 37°C for 24 h. Thereafter, culture media was aspirated and replaced with 5 mg/ml of MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)] solution. After 3 h of incubation, isopropanol was added for solubilizing formazan produced by viable cells and absorbance was recorded at 570 nm using a microplate reader (SPECTROstar nano, BMG Labtech, Germany). The viability was calculated as – % of cell viability= [100-{(C –T)/ C X100}], where “C’’ is the mean optical density of control (without treatment) and ‘’T’’ is the mean optical density of cells treated with drug.
Trypan blue exclusion assay
To the rapidly growing WRL-68 and Hep-G2 cell line, culture media was replaced with PBS as described earlier and predetermined IC50 dose of 5-FU (300µM conc. for HepG2 and 250 µM conc. for WRL-68) was applied in each mentioned condition. After 3 h of incubation, cells were scrapped and centrifuged at 2000 rpm for 5 min. The resultant pellet was then restored in fresh 1X PBS. At last, cell suspension and trypan blue solution (0.04%) were mixed in a 1:1 ratio and the number of viable and non-viable cells were quantified51 using hemocytometer under a compound microscope (Olympus CK40).
Morphological assessment assay
Morphological differences were identified under the following conditions – control (cells with complete media), control + 5-FU, PBS and PBS + 5-FU. Visualization was achieved under the 20X objective of a phase contrast microscope (Olympus CK40).
Acidic vacuole organelle detection (AVO) by acridine orange staining
AVOs formation in WRL-68 and HepG2 cells were detected by staining the cells with 2 µg/ml acridine orange solution for 15 min in dark52. Here, 5-FU was added in combination with PBS to determine a comparative autophagy induction. Rapamycin-treated cells were taken as positive control (5 µM). The prepared slides were visualized under 40X magnification of a fluorescence microscope (Magnus MLXi, India).
Determination of superoxide formation
Cells were treated with 5-FU in nutrient-sufficient and deficient conditions and production of intracellular superoxide formation was detected by staining the cells with DHE (dihydroethidium) dye for 1 h in dark30. Visualization was performed with a fluorescence microscope (Magnus MLXi, India) under 40X magnification.
Qualitative and quantitative measurement of lipidation
Lipidation status was checked by oil red o staining53. For visualization purposes, the treated and untreated cells were first fixed by using 10% formalin. Then, the fixed cells were stained with oil red o dye for 20 min in dark and visualized under 40X magnification of a compound microscope (Olympus, USA). Another set was also prepared for quantitative measurement of lipid accumulation according to the manufacturer’s protocol53. The amount of stain retained by cells was extracted with 100% isopropanol and absorbance was recorded at 492 nm by SPECTROstar nano plate reader (BMG Labtech, Germany). 100% isopropanol was used as background control to subtract the background signal.
Apoptosis determination via AnnexinV-FITC and propidium iodide (PI) staining
Apoptosis was examined by AnnexinV-FITC and Propidium Iodide staining assay54,55. Briefly, cell pellets were suspended in 100 µl of 1X binding buffer. Following that, 1 µl of AnnexinV-FITC and 2 µl of PI were added and the resultant mixture was incubated for 10 min at room temperature in dark. Fluorescence microscopic (Magnus MLXi, India) observations at 480/530 nm were then recorded.
Mitochondrial membrane damage assay by TMRE (tetramethylrhodamine ethyl ester) staining
Determination of mitochondrial membrane damage was achieved by staining the cells with TMRE (50 nM TMRE dissolved in PBS) dye for 20 min56. Similar treatment conditions were considered and emitted fluorescence was recorded by fluorescence microscopic observation (Magnus MLXi, India) under the 40X objective lens.
Cell death assessment via acridine orange/ethidium bromide (AO/EtBr) staining
To evaluate cell death by 5-FU under an autophagic environment, differential acridine orange/ethidium bromide staining was performed48. All cells were stained with 2 µg/ml of AO/EtBr solution for 15 min in dark. Then, slides were prepared and viewed under a fluorescence microscope (Magnus MLXi, India).
Statistical analysis
All values are represented as mean ± SD of three biological replicates. All the Graphs were prepared using GraphPad Prism v10.0.1 (GraphPad Software, San Diego, California). Significance was evaluated by one-way ANOVA followed by Tukey’s post hoc test for multiple comparisons between control and treatments. Differences were considered significant with p < 0.05.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
-
McGlynn, K. A., Petrick, J. L. & El-Serag, H. B. Epidemiology of hepatocellular carcinoma. Hepatol. 73, 4–13 (2021).
Google Scholar
-
World Health Organization, International agency for research on cancer. (2019).
-
Farshid, P. et al. Repetitive chemoembolization of hypovascular liver metastases from the most common primary sites. Future Oncol. 9, 419–426 (2013).
Google Scholar
-
Penchev, D. K., Vladova, L. V., Zashev, M. Z. & Gornev, R. P. Distant liver metastases as a major factor influencing survival in patients with colorectal Cancer. Folia Med. 58, 182 (2016).
Google Scholar
-
Xu, T. et al. Synergistic effects of Curcumin and 5-Fluorouracil on the Hepatocellular Carcinoma in vivo and vitro through regulating the expression of COX-2 and NF-κB. J. Cancer. 11, 3955 (2020).
Google Scholar
-
Wu, X. L., Chen, Y., Kong, W. C. & Zhao, Z. Q. Amyloid precursor protein regulates 5-fluorouracil resistance in human hepatocellular carcinoma cells by inhibiting the mitochondrial apoptotic pathway. J. Zhejiang Univ. Sci. B. 21, 234–245 (2020).
Google Scholar
-
Wei, L. et al. The emerging role of microRNAs and long noncoding RNAs in drug resistance of hepatocellular carcinoma. Mol. Cancer. 18, 1–11 (2019).
Google Scholar
-
Ge, J. et al. Upregulation of autophagy-related gene-5 (ATG-5) is associated with chemoresistance in human gastric cancer. PloS One. 9, e110293 (2014).
Google Scholar
-
Liu, H., He, Z. & Simon, H. U. Targeting autophagy as a potential therapeutic approach for melanoma therapy (In Seminars in cancer biology) Vol. 23, No. 5, 352–360Academic Press, (2013).
-
Ma, L. & Wang, Y. JAK2/STAT3 inhibitor reduced 5-FU resistance and autophagy through ATF6-mediated ER stress. J. Recept Sig Transd. 42, 206–213 (2022).
Google Scholar
-
Khan, S. U., Fatima, K., Aisha, S. & Malik, F. Unveiling the mechanisms and challenges of cancer drug resistance. Cell. Commun. Signal. 22, 109 (2024).
Google Scholar
-
Deng, L. F. et al. Anti-proliferation effect of sorafenib in combination with 5-FU for hepatocellular carcinoma in vitro: antagonistic performance and mechanism. Chin. J. Hepatol. 21, 845–849 (2013).
-
Boueroy, P. et al. Synergistic effect of Forbesione from Garcinia Hanburyiin Combination with 5-Fluorouracil on Cholangiocarcinoma. Asian Pac. J. Cancer Prev. 18, 3343 (2017).
Google Scholar
-
Ohsumi, Y. Historical landmarks of autophagy research. Cell. Res. 24, 9–23 (2014).
Google Scholar
-
Khan, S. U., Fatima, K. & Malik, F. Understanding the cell survival mechanism of anoikis-resistant cancer cells during different steps of metastasis. Clin. Exp. Metastasis. 39, 715–726 (2022).
Google Scholar
-
Jain, M. V. et al. Interconnections between apoptotic, autophagic and necrotic pathways: implications for cancer therapy development. J. Cell. Mol. Med. 17, 12–29 (2013).
Google Scholar
-
Chude, C. I. & Amaravadi, R. K. Targeting autophagy in cancer: update on clinical trials and novel inhibitors. Int. J. Mol. Sci. 18, 1279 (2017).
Google Scholar
-
Ichimiya, T. et al. Autophagy and autophagy-related diseases: a review. Int. J. Mol. Sci. 21, 8974 (2020).
Google Scholar
-
Nazim, U. M. et al. Activation of autophagy flux by metformin downregulates cellular FLICE–like inhibitory protein and enhances TRAIL-induced apoptosis. Oncotarget. 7, 23468 (2016).
Google Scholar
-
Bagamanshina, A. V. et al. Cytotoxic and antitumor activity of lactaptin in combination with autophagy inducers and inhibitors. BioMed Res. Int. 4087160 (2019). (2019).
-
Van Niekerk, G., Hattingh, S. M. & Engelbrecht, A. M. Enhanced therapeutic efficacy in cancer patients by short-term fasting: the autophagy connection. Front. Oncol. 6, 242 (2016).
Google Scholar
-
de Groot, S., Pijl, H., van der Hoeven, J. J. & Kroep, J. R. Effects of short-term fasting on cancer treatment. J. Exp. Clin. Cancer Res. 38, 1–14 (2019).
Google Scholar
-
Omar, E. M., Omran, G. A., Mustafa, M. F. & El-Khodary, N. M. Intermittent fasting during adjuvant chemotherapy may promote differential stress resistance in breast cancer patients. J. Egypt. Natl. Canc Inst. 34, 38 (2022).
Google Scholar
-
Koppold-Liebscher, D. et al. Short-term fasting accompanying chemotherapy as a supportive therapy in gynecological cancer: protocol for a multicenter randomized controlled clinical trial. Trials. 21, 1–12 (2020).
Google Scholar
-
de Gruil, N., Pijl, H., van der Burg, S. H. & Kroep, J. R. Short-term fasting synergizes with solid cancer therapy by boosting antitumor immunity. Cancers. 14, 1390 (2022).
Google Scholar
-
Liu, F. et al. LncRNA NEAT1 knockdown attenuates autophagy to elevate 5-FU sensitivity in colorectal cancer via targeting miR‐34a. Cancer Med. 9, 1079–1091 (2020).
Google Scholar
-
Amaravadi, R., Kimmelman, A. C. & White, E. Recent insights into the function of autophagy in cancer. Genes Dev. 30, 1913–1930 (2016).
Google Scholar
-
Antunes, F., Pereira, G. J., Jasiulionis, M. G., Bincoletto, C. & Smaili, S. S. Nutritional shortage augments cisplatin-effects on murine melanoma cells. Chem. Biol. Interact. 281, 89–97 (2018).
Google Scholar
-
Kim, H. K. et al. Academic Press,. Current and upcoming mitochondrial targets for cancer therapy (In Seminars in Cancer Biology) Vol. 47, 154–167 (2017).
-
Ali, S. et al. β-cyclodextrin-stabilized biosynthesis nanozyme for dual enzyme mimicking and Fenton reaction with a high potential anticancer agent. ACS Omega. 7, 4457–4470 (2022).
Google Scholar
-
Pajaud, J., Kumar, S., Rauch, C., Morel, F. & Aninat, C. Regulation of signal transduction by glutathione transferases. Int. J. Hepatol. 137676 (2012). (2012).
-
Klionsky, D. J. et al. Autophagy in major human diseases. EMBO J. 40, e108863 (2021).
Google Scholar
-
Anton, S. D. et al. Flipping the metabolic switch: understanding and applying the health benefits of fasting. Obesity. 26, 254–268 (2018).
Google Scholar
-
Dutta, A., Thakur, S., Dey, D. K. & Kumar, A. Cisplatin and starvation differently sensitize Autophagy in Renal Carcinoma: a potential therapeutic pathway to target variegated drugs resistant cancerous cells. Cells. 13, 471 (2024).
Google Scholar
-
Murphy, M. P. et al. Guidelines for measuring reactive oxygen species and oxidative damage in cells and in vivo. Nat. Metabol. 4, 651–662 (2022).
Google Scholar
-
Kang, R., Zeh, H. J., Lotze, M. T. & Tang, D. J. C.D. The beclin 1 network regulates autophagy and apoptosis. Cell. Death Dif. 18, 571–580 (2011).
Google Scholar
-
Patterson, R. E. et al. Intermittent fasting and human metabolic health. J. Acad. Nutr. Diet. 115, 1203 (2015).
Google Scholar
-
Tripathi, D. N. et al. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2–mediated suppression of mTORC1. Proc. Natl. Acad. Sci. 110, E2950-E2957 (2013).
-
Khan, S. U., Fatima, K., Malik, F., Kalkavan, H. & Wani, A. Cancer metastasis: molecular mechanisms and clinical perspectives. Pharmacol. Ther. 250, 108522 (2023).
Google Scholar
-
Tian, R., Seim, I., Ren, W., Xu, S. & Yang, G. Contraction of the ROS scavenging enzyme glutathione S-transferase gene family in cetaceans. G3: Genes Genome Genet. 9, 2303–2315 (2019).
Google Scholar
-
Liu, J. et al. Nitric oxide interacts with caveolin-1 to facilitate autophagy-lysosome-mediated claudin-5 degradation in oxygen-glucose deprivation-treated endothelial cells. Mol. Neurobiol. 53, 5935–5947 (2016).
Google Scholar
-
Zhang, J., Deng, Y. & Khoo, B. L. Fasting to enhance Cancer treatment in models: the next steps. J. Biomed. Sci. 27, 58 (2020).
Google Scholar
-
de Groot, S. et al. Fasting mimicking diet as an adjunct to neoadjuvant chemotherapy for breast cancer in the multicentre randomized phase 2 DIRECT trial. Nat. Commun. 11, 3083 (2020).
Google Scholar
-
Steinhauser, M. L. et al. The circulating metabolome of human starvation. JCI Insight, 3, (2018).
-
Rennert, C. et al. The diurnal timing of starvation differently impacts murine hepatic gene expression and lipid metabolism–a systems biology analysis using self-organizing maps. Front. Physiol. 9, 1180 (2018).
Google Scholar
-
Guo, S. et al. A rapid and high content assay that measures cyto-ID-stained autophagic compartments and estimates autophagy flux with potential clinical applications. Autophagy. 11, 560–572 (2015).
Google Scholar
-
Yan, J. et al. Nur77 attenuates inflammatory responses and oxidative stress by inhibiting phosphorylated IκB-α in Parkinson’s disease cell model. Aging. 12, 8107 (2020).
Google Scholar
-
Murugan, S. & Amaravadi, R. K. Methods for studying autophagy within the tumor microenvironment. Tumor Microenvironment: Study Protocols 145–166 (2016).
-
Oh, S. H., Choi, Y. B., Kim, J. H., Weihl, C. C. & Ju, J. S. Quantification of autophagy flux using LC3 ELISA. Anal. Biochem. 530, 57–67 (2017).
Google Scholar
-
Denizot, F. & Lang, R. Rapid colorimetric assay for cell growth and survival: modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods. 89, 271–277 (1986).
Google Scholar
-
Strober, W. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. 21, A-3B (1997).
-
Thomé, M. P. et al. Ratiometric analysis of Acridine Orange staining in the study of acidic organelles and autophagy. J. Cell. Sci. 129, 4622–4632 (2016).
Google Scholar
-
Yan, Q. et al. Autophagy activation contributes to lipid accumulation in tubular epithelial cells during kidney fibrosis. Cell. Death Discov. 4, 39 (2018).
Google Scholar
-
Rieger, A. M., Nelson, K. L., Konowalchuk, J. D. & Barreda, D. R. Modified annexin V/propidium iodide apoptosis assay for accurate assessment of cell death. J. Vis. Exp. 50, e2597 (2011).
-
Koç, E., Çelik-Uzuner, S., Uzuner, U. & Çakmak, R. The detailed comparison of cell death detected by annexin V-PI counterstain using fluorescence microscope, flow cytometry and automated cell counter in mammalian and microalgae cells. J. Fluoresc. 28, 1393–1404 (2018).
Google Scholar
-
Crowley, L. C., Christensen, M. E. & Waterhouse, N. J. Measuring mitochondrial transmembrane potential by TMRE staining. Cold Spring Harb. Protoc. 087361 (2016). (2016).
Acknowledgements
Authors sincerely thank, West Bengal State Government as one of the author; Ankita Dutta received fellowship through Swami Vivekananda Merit-cum-Means Scholarship (SVMCM) (Applicant ID WBP191578382293).
Author information
Authors and Affiliations
Contributions
A.K. and A.D. conceived and designed the research. A.D. conducted all experiments and wrote the original manuscript. A.C. and T.G. collected data. A.K. supervised, reviewed and edited the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
Reprints and permissions
About this article
Cite this article
Dutta, A., Chakraborty, A., Ghosh, T. et al. 5-Fluorouracil induces apoptosis in nutritional deprived hepatocellular carcinoma through mitochondrial damage.
Sci Rep 14, 23387 (2024). https://doi.org/10.1038/s41598-024-73143-y
-
Received: 24 June 2024
-
Accepted: 13 September 2024
-
Published: 08 October 2024
-
DOI: https://doi.org/10.1038/s41598-024-73143-y
Keywords
- Hepatocellular carcinoma
- 5–Fluorouracil
- Starvation
- Autophagy
- Reactive oxygen species (ROS)
- Apoptosis
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.