Abstract
All life forms across the globe are experiencing drastic changes in environmental conditions as a result of global climate change. These environmental changes are happening rapidly, incur substantial socioeconomic costs, pose threats to biodiversity and diminish a species’ potential to adapt to future environments. Understanding and monitoring how organisms respond to human-driven climate change is therefore a major priority for the conservation of biodiversity in a rapidly changing environment. Recent developments in genomic, transcriptomic and epigenomic technologies are enabling unprecedented insights into the evolutionary processes and molecular bases of adaptation. This Review summarizes methods that apply and integrate omics tools to experimentally investigate, monitor and predict how species and communities in the wild cope with global climate change, which is by genetically adapting to new environmental conditions, through range shifts or through phenotypic plasticity. We identify advantages and limitations of each method and discuss future research avenues that would improve our understanding of species’ evolutionary responses to global climate change, highlighting the need for holistic, multi-omics approaches to ecosystem monitoring during global climate change.
Introduction
The impact of global climate change (GCC) is affecting all forms of life across all biomes, with potentially detrimental consequences to the persistence of species and ecosystem functioning1,2,3,4,5,6. Shifts in environmental conditions, such as warming temperatures and ocean acidification, have direct effects on organisms by challenging physiological limits7,8,9 or altering phenology10,11. Indirect effects of climate change can also threaten biodiversity by increasing the spread of novel pathogens and severity of disease outbreaks12, or by introducing new species that alter predation and competition dynamics in communities13.
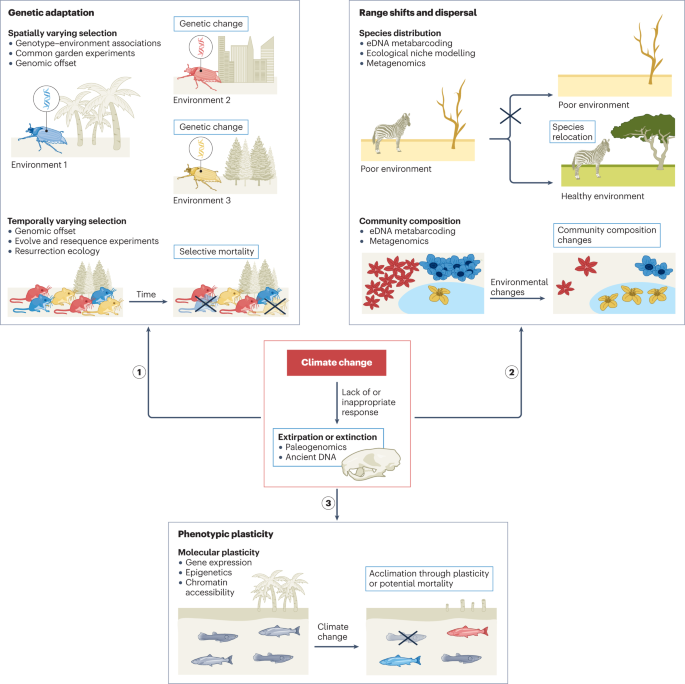
The rapid pace of GCC may preclude adequate responses in many populations, thus increasing the rate of decline and extinction14. Extinction can be avoided by species shifting their geographical distribution to more favourable habitats, acclimatizing to stressful conditions through phenotypic plasticity — the ability of one genotype to express different phenotypes in different environments — or adapting through genetic change (Fig. 1). However, it is challenging to understand when eco-evolutionary responses will occur and to differentiate between the aforementioned responses to identify potential evolutionary ‘winners’ and ‘losers’, such as species with low adaptive capacity6,15,16,17. Many studies have attributed phenotypic changes in natural populations to GCC-induced phenotypic plasticity3,5,18. However, plasticity and genetic effects are difficult to disentangle as both contribute to phenotype, and plastic mechanisms are often partially genetically controlled19,20,21. Genomic data need to be supplemented with experiments in controlled conditions to document evolutionary change over time and by integrating other omics approaches, such as transcriptomics and epigenomics, to adequately infer the genetic versus plastic basis of responses to GCC3. Monitoring temporal genomic and phenotypic changes drastically improves our understanding of how GCC affects organisms; although this approach presents logistical issues for many species, it is becoming increasingly feasible with recent omics technologies.

Global climate change (GCC) poses a significant threat to species, although they can adapt genetically through spatially or temporally varying selection (response 1), cope through range shifts and dispersal when possible to avoid extirpation (response 2) or acclimate to GCC through phenotypic plasticity (response 3). Various methods that integrate genomic and/or epigenomic tools (listed as bullet points) can be used in both natural environments and experimental laboratory conditions to assess how species are responding to GCC (Table 1). Lack of or inappropriate responses can result in extirpation or extinction (red box), which can also inform on the historical effects of GCC on species and communities. eDNA, environmental DNA.
Next-generation sequencing methods have transformed the field of population and functional genomics over the past decade by enabling the screening of whole-genome variation within and between species across space and time22. This makes it possible to characterize the genome-wide architecture that underlies adaptive traits and the genome-wide response to selection induced by natural or anthropogenic factors, including GCC6. New analytical methods can forecast range shifts under predicted climate change for individuals adapted to different climatic conditions and determine the potential for rescue effects to support population persistence. Moreover, genome-wide plastic responses to new environmental conditions through changes in gene expression and epigenetic modifications can be investigated using molecular approaches, in both controlled conditions and natural environments. Recent developments in metagenomics and metabarcoding also represent powerful methods to monitor community composition of all species under GCC. Together, this array of omics technologies presents a powerful toolbox to understand the effects of GCC on species and communities.
Here, we review the application of methods used in recent years to infer or predict organismal responses to GCC, in both natural and experimental conditions (Table 1). For a comprehensive review of potential evolutionary responses to GCC23 or the technical details of the methods described24,25,26, we refer readers to other reviews. Our aim is to summarize and highlight the broad range of applications of diverse methodologies over the past decade in the context of GCC, using key examples. We present advantages and limitations associated with the reviewed methods (Table 1), as well as future perspectives for improving predictions of the multifaceted responses to GCC across systems.
Identifying adaptive genetic variation
Characterization of spatial patterns of adaptive genetic variation in wild populations is an important first step towards understanding the potential for adaptation to GCC. Spatially varying selection can drive local adaptation and maintain standing genetic variation, facilitating adaptive responses to environmental change. For example, one study identified physiological differences and genomic divergence despite high gene flow between Acropora tenuis populations occupying different habitats in an isolated coral reef system in Western Australia27. This finding supports a role for selection driven by spatially varying environmental conditions to maintain genetic variation that confers resilience to heat stress in habitats experiencing more extreme conditions.
Population genomic methods, such as genome scans for signals of divergent selection, are now extensively used to identify candidate adaptive genomic variants, typically SNPs. For example, differentiation-based scans detect candidate adaptive loci on the basis of signals of locus-specific genetic differentiation between populations that deviate from expectations under a neutral model (for example, FST outliers) and are thus presumably subject to selection28. This approach has been used to identify highly differentiated SNPs across genetically distinct populations that were subsequently linked with bioclimatic variables such as temperature and salinity and functional genes potentially involved in thermal tolerance or salinity stress response mechanisms29. Other genome scan approaches that directly incorporate environmental variables, known as genotype–environment associations (GEAs)28,30,31, detect candidate adaptive loci on the basis of direct statistical associations between allele frequencies and environmental variables and can identify key climatic factors that are driving local adaptation across heterogeneous landscapes32,33,34,35. Several GEA methods exist and their performance under various scenarios has been reviewed elsewhere36,37,38. Most conventional GEA methods use linear models to test genetic–environment relationships30, although methods such as regression tree-based models can capture nonlinear relationships and have been used to detect genomic variants associated with climate adaptation39,40. Multivariate methods, especially redundancy analysis (RDA), are commonly used to identify GEAs given their ability to model multi-dimensional relationships and detect weak polygenic signatures of selection compared with univariate methods37,41. Window-based approaches have also been applied to characterize the genetic basis of local adaptation and have shown promising levels of performance when compared with other GEA methods42.
Understanding spatial patterns of climate-associated genetic diversity can provide insight into the adaptive potential of populations43 and is a common focus of landscape genomics studies. Detection of local adaptation to environmental conditions using GEAs combined with assessments of dispersal capacity estimated with landscape resistance suggests that the potential for evolutionary responses to changing conditions may be limited in some populations, as documented in the streamside salamander Ambystoma barbouri44. When coupled with an ecological niche model (ENM), the spatial distribution of climate-adapted loci can be related to range-wide environmental suitability, thereby identifying regions that may be sources of adaptive genetic variation to unfavourable or changing conditions at niche limits45 (Fig. 2). Accordingly, studies using GEAs provide essential information for implementing strategies aimed at protecting species that may be vulnerable to effects of GCC46, spatial planning that incorporates adaptive genetic variation in delineating conservation units or prioritizing areas for protection47,48, and informing actions such as assisted gene flow to enhance the adaptive potential of small populations49,50.

a, Aguirre-Liguori et al. sampled populations of teosinte (Zea mays mexicana) along an environmental gradient in southern Mexico45. b, Environmental layers and species occurrence data were combined to predict the species’ niche distribution using an ecological niche model. c, Candidate SNPs were detected by performing genome scans of genotype–environment associations (GEAs). d, Using the predicted niche distribution, the authors defined the niche centroid based on the mean value of each environmental axis and calculated an environmental distance (d) between each population and the niche centroid; populations with a higher d inhabit more unsuitable (niche edge) habitats. e, For each candidate SNP, the authors fitted an environmental cline and identified populations in which the SNP is likely to be adaptive (filled circles; α) versus populations in which the SNP evolves neutrally (open circles). f, The authors detected a significant positive correlation between the number of adaptive SNPs per population and the distance of each population from the niche centroid (d). This finding suggests that populations that occur at niche limits may be important sources of adaptive variation for populations experiencing environmental change. Parts c, e and f adapted with permission from ref. 45, Wiley.
Signatures of selection may be confounded by neutral population structure and spatial autocorrelation with environmental factors, leading to incorrect inference of local adaptation from GEAs (and differentiation-based genome scans)28. Many GEA approaches incorporate corrections to account for the effects of demography and geography28. RDA can also be adapted to include conditioning variables using a partial RDA (pRDA), enabling variance partitioning between different sets of predictors (for example, environmental, spatial or demographic) and correcting for confounded effects41. For example, after incorporating spatial variables representing dendritic riverine network structure, 4% of genetic variation in the Australian rainbowfish (Melanotaenia fluviatilis) populations was attributed to the environment, and >700 loci exhibited associations with temperature, precipitation and stream flow34. Although this approach reduces the number of false-positive detections, it can also exclude true positives when environmental gradients coincide with spatial or demographic processes37,51.
Although GEAs are powerful and convenient approaches for discovering climate-associated genetic variation, there are some recognized limitations. The development of GEA methods relies on the basic assumption that adaptive alleles will form frequency clines associated with the selective environment. However, using a range of simulations, a recent study showed that nonclinal allele frequency patterns can evolve despite phenotypic clines associated with climatic gradients, which may lead to incorrect inference of adaptive SNPs based on GEAs51. Moreover, causal variants, especially those of small effect, may not be readily detectable52, and the predictability of the genomic selection response for polygenic traits depends on an understanding of the underlying adaptive architecture (including linkage disequilibrium, epistasis and recombination rate variation)53,54,55. Despite the limitations of GEA, an extension to the RDA framework has been shown to accurately predict multivariate quantitative traits from landscape genomic data across a range of demographic and selection scenarios and complex genomic architectures51. Finally, additional evidence is needed to corroborate genetic–environment associations and their involvement in climate adaptation, including experimental approaches that provide evidence of fitness differences associated with candidate variants56,57. For example, in common garden experiments, individuals from different environments are reared in a common environment to elucidate the genetic and environmental components that underlie phenotypic differences. Alternatively, reciprocal transplants can be performed, whereby individuals originating from each of two (or more) environments are introduced into the other environment to characterize local adaptation based on fitness differences between individuals reared in home versus away environments, or between resident and immigrant individuals in a given environment. Although this type of validation is not always feasible, the outcomes of many studies support the broader application of GEAs for understanding population-level patterns of climate adaptation31,49.
Genomic vulnerability to GCC
Genomic offset (also known as genetic offset58 or genomic vulnerability59) combines genomic and environmental data from different time points and/or locations to assess the degree of possible maladaptation to new environmental conditions. It is an increasingly popular approach for predicting how populations or species will respond to GCC and was inspired by genomic selection methods that aim to predict phenotype based on genotype. This approach is classically a two-step process: first, GEA approaches identify putatively adaptive variation; and second, spatial and/or temporal extrapolation typically relates current patterns of adaptive genomic composition of populations to climate. The predicted optimal population genomic compositions are projected across a species’ range (space), onto future climatic conditions (time), or both, to estimate the magnitude of genetic shift (in allele or genotype frequencies) required by populations to maintain the current fitness status quo under different climates using a modelling approach similar to ENMs (Fig. 3). Several models have been developed to estimate genomic offset statistics (reviewed elsewhere5,60,61). Most studies that aim to predict future genomic (mal)adaptation of populations have used the Gradient Forest (GF) method58,59,62,63,64,65. GF was originally developed to model spatial variation in community composition and subsequently used on SNP data to model turnover in allele frequencies and estimate the genetic offset that climate change would induce for balsam poplar (Populus balsamifera) populations across North America58. Generalized dissimilarity modelling (GDM)66 is the second community-level modelling method adapted to extrapolate genetic–environment relationships and can be used to predict genetic differentiation (FST) between contemporary and future populations as a function of the environmental distance between current and future climatic conditions63,67. RDA has also been used to produce spatial extrapolations of intraspecific adaptive genetic variation68, predict adaptive genotypes for reforestation sites69 and spatially predict genomic offset associated with climate change41. Risk of nonadaptedness (RONA)70 uses a linear regression (at a single locus level) in previously identified, putatively adaptive loci without correction for population structure to estimate the expected allele frequency required under the new environmental conditions. Pinas-Martins et al.71 expanded RONA by using a weighted average R2 of the regression to investigate the adaptation potential of cork oak to survive predicted climate change. The spatial areas of genotype probability (SPAG) method uses multivariate logistic regressions to compare the adaptive landscape under current and future climatic conditions72. Recently, the ‘genetic gap’ was proposed as a geometric measure of genomic offset that unifies previously proposed genomic offset statistics in a common framework73. Considering a duality between genetic and environmental space, a theoretical framework was developed that linked genomic offset statistics to a non-Euclidean geometry of the ecological niche.

a, Based on the Zea mays mexicana SNP data set (Fig. 2 and ref. 45), Aguirre-Liguori et al.75,76 first used the current adaptive genetic turnover identified between two geographically and genetically distinct clusters and significantly associated with temperature. b, The authors then projected the geographical range of the two genetic clusters in the present and future (2070) conditions using Maxent228 and the WorldClim database229. c, A Gradient Forest model was used to assess genomic offset in each sampled population. The estimated genomic offset combined across all populations within the colder southern cluster (blue) and warmer northern cluster (red) indicated that the predicted genomic offset is higher on average for southern populations. d, Maps of potential migration using the present and future distribution models for putative warm-adapted alleles were constructed to determine landscape resistance as a proxy of limitations to successful migration using circuit theory230. Comparison between populations suggested that some populations have few dispersal routes. e, Adaptive and maladaptive gene flow between populations from the two clusters were inferred using coalescent simulations231 to assess whether the population receives an influx of warmer-adapted alleles from northern populations or is swamped with maladaptive alleles from the south. f, The presence of genomic load, that is, genetic variants putatively reducing the fitness of a population relative to a local fitness optimum, was investigated via a measure that compared the proportion and population frequency of non-synonymous SNPs with those of synonymous SNPs. g, The findings indicate that southern populations have higher estimated loads than northern ones. h, Moreover, for both clusters there is a positive and significant trend between genomic load and distance from the niche centroid (d in Fig. 2). This effect is more pronounced for northern populations, suggesting that they are particularly likely to be subjected to evolutionary forces that produce genetic load. With this integrated framework, the authors highlighted the importance of taking into consideration the full ‘give and take’ between adaptation and potentially transgressing forces such as gene flow, dispersal and load to make accurate predictions of population vulnerability under global climate change (GCC) (particularly about the fate of small populations at the edge of a species’ geographical range). Parts a, c–h adapted from ref. 75, Springer Nature Limited.
The proposed measures of genomic offset are empirically validated but also have well-identified limitations61,74,75 (Table 1). Such analyses are stronger when incorporated with other approaches. Aguirre-Liguori et al.75,76 combined genetic offset analysis with other approaches in a study on synthesized risk assessments of teosinte (Zea mays parviglumis) populations experiencing GCC (Fig. 3). In a study of yellow warbler (Setophaga petechia), a combination of genomic offset scores and demographic population trends across the species’ breeding range showed that populations that require the greatest shifts in allele frequencies to keep pace with future climate change have experienced the largest population declines, suggesting that failure to adapt may already be having negative effects59. Across an elevational gradient in the Australian Wet Tropics, Brauer et al.77 showed that hybrid populations between a widespread generalist and several narrow-range endemic species exhibited reduced genomic vulnerability to projected climates compared with pure narrow endemic species. This study goes further than previous studies by both considering genomic vulnerability and providing empirical evidence for gene flow mitigating maladaptation. The combination of genomic offset scores and phenotypic data (flowering time) of the pearl millet (Pennisetum glaucum), a nutritious staple cereal cultivated in arid and low-fertility soils in sub-Saharan Africa, predicts that the most vulnerable areas will benefit from using landraces that already grow in equivalent climate conditions today78.
Investigating temporal evolutionary change
Evolve and resequence experiments
Experimental evolution allows the direct study of evolution through controlled manipulations of organisms over many generations and represents the most rigorous approach for understanding trait evolution due to different environmental conditions79,80,81. As many organisms have prohibitively long generation times, most studies focus on species that reproduce rapidly, such as microorganisms. Pioneering experimental evolution studies using Escherichia coli as a model system studied evolutionary forces and their impact on trait evolution under controlled conditions over 75,000 generations82,83,84,85,86,87. Evolve and resequence (E&R) provides a powerful means to track molecular evolution in ‘real time’ and dissect the adaptive architecture of selected traits at the highest genomic level of resolution88,89,90 (Fig. 4). Whole-genome sequencing is becoming more affordable and prevalent in E&R experiments, although many experiments on microbial organisms or small multicellular organisms with very short generation times still use Pool-seq to sequence pools of many individuals, which is cost-effective and yields accurate genome-wide allele frequency estimates90,91. Numerous E&R experiments, mostly using Drosophila, have investigated evolutionary issues pertaining to GCC, including the role of standing genetic variation versus de novo mutations in adaptation92, the synergistic effects of different stressors and population ancestry93,94 and the role of genetic adaptation versus plasticity in response to high temperatures95,96. Although E&R studies are typically conducted over many generations, several studies have shown that adaptive genetic variation can be identified with a single or a few generations of selection97,98,99,100.

Deatherage et al.232 investigated the specificity of genome evolution in Escherichia coli at various temperatures. a, Thirty populations were evolved for 2,000 generations, with six replicates in each of five different thermal regimes. The genomes were sequenced at one end point from each population and compared with the ancestral population. The authors used the breseq program233 to identify mutations in evolved genomes. b, Summary of the 159 derived mutations observed in the 30 sequenced genomes by the type of genetic change. Across all populations, 159 de novo mutations were found. Most mutations (57%) were single base substitutions, 87% of which were non-synonymous substitutions or nonsense substitutions, which provided a strong signal of adaptive evolution. The other 43% of the mutations in the temperature-evolution experiment (TEE) lines comprised small insertions or deletions (indels), large deletions, amplifications and rearrangements. Populations that evolved under the same thermal regime exhibited four times more overlap (17% versus 4%) in which genes were mutated compared with those evolved at different temperatures. c, Genes (rows) affected by at least two qualifying mutations in the 30 TEE clones (columns, organized by temperature regime and populations –1, +1, –2, +2, –3 or +3). Results revealed a clear signal of genomic specificity in how populations adapted to different temperature regimes whereby mutations converged on a distinctive set of genes with signature mutations in each treatment. The figure presents the genes containing at least two qualifying mutations. Five ‘signature’ mutations, shown as coloured squares, are significantly associated with one or two treatments. One of the populations that was evolved at 37 °C for another 18,000 generations accumulated mutations in signature genes that were strongly associated with adaptation to the other temperature regimes. This landmark study demonstrates that the genomic signature of adaptation is highly specific, although populations evolving under the different regimes might eventually show more genetic parallelism than divergence234. Parts a–c adapted with permission from ref. 232, PNAS.
Despite their many benefits, it is unclear how E&R results relate to processes of adaptation in nature79. This was recently investigated by comparing patterns of gene expression divergence between populations of Drosophila melanogaster adapted for 80 laboratory generations to two distinct temperature regimes with those identified by contrasting natural populations across two different latitudinal clines101. The authors found that 203 genes in seven co-expression modules evolved temperature-specific expression changes in the laboratory populations. Moreover, their results revealed a positive correlation in temperature-induced expression between laboratory and natural populations from the two clines. They concluded that well-designed E&R experiments can inform how populations respond to selection in natural environments, although another recent study on the harlequin fly (Chironomus riparius) concluded that results from E&R experiments could not provide insights into thermal adaptation potential in natural populations102.
Resurrection ecology and genomics
Resurrection ecology provides information on the phenotypic variation and fitness of past organisms by resurrecting dormant life stages, for example, seeds, eggs, cysts or spores that accumulate in lacustrine and marine sediments, soil or ice, allowing insight and direct study of organisms from before our lifetime79. Resurrection ecology is a powerful approach79,103 that predates the ‘genomic era’104,105,106, yet few studies on the impact of GCC on natural populations have incorporated genomics into resurrection ecology studies107 despite tremendous advantages108,109,110,111,112,113. Temporally stratified propagule banks can be accurately dated such that population genomic and phenotypic evolution can be aligned with temporal environmental changes103. Resurrection ecology combined with genomics can also assess the role of new mutations versus standing genetic variation in adaptive phenotypic responses to environmental changes114,115,116.
The best examples of combination of resurrection ecology with genomics come from gene expression studies, including a key study on the aquatic crustacean Daphnia magna117 (Fig. 5). Another study in the common mustard Brassica rapa examined evolutionary changes in gene expression in seeds from two populations over 18 years during which precipitation fluctuated dramatically118. By comparing gene expression of ancestors and descendants of these resurrected populations grown under common conditions, they found that hundreds of genes associated with drought stress and flowering time showed transcriptional differences between ancestors and descendants of the two populations. This study, along with others119,120, indicates that evolutionary changes in gene regulation may provide rapid adaptive responses to contemporary shifts in climatic conditions.

Jansen et al.235 used a candidate gene approach to document evolutionary response of gene expression in a resurrected population of Daphnia magna that experienced changes in temperature over the past 40 years and from contemporary (experimental) populations differing in thermal tolerance. a, Sediments from Felbrigg Hall Lake, Norfolk, UK, were sampled and dated, and dormant eggs were hatched and cultured from a colder and a warmer period. b, The upper 2 cm of the sediment from mesocosms exposed to either ambient or ambient + 4 °C temperature treatment was sampled for resting eggs, and hatched D. magna were used for candidate gene analysis. c, Reaction norms for differential expression were measured at the candidate genes between heat treatment and control. Authors documented evolutionary changes in gene expression between warm and cold-adapted populations and assessed evolutionary response to temperature changes. They used model-averaged effect sizes to quantify the impact of each term and applied a false coverage ratio correction to correct for multiple testing before assessing the significance of each term. Most of the tested genes (57%, a subset of 12 genes illustrated here) in the contemporary populations showed a plastic response to heat treatment compared with only 23% in the resurrected population. These results thus indicated that most genes apparently lost plasticity of expression in the face of evolutionary (thermal) constraints in the natural (resurrected) population. Their study also identified candidate genes likely linked to thermal adaptation. Figure adapted with permission from ref. 235, Wiley.
Evolutionary change and time series
Temporal evolutionary changes can also be investigated at the genome level from time series data5,121,122. This involves sampling natural populations, using historical samples123,124 to measure allele frequency at various times, correlate genotypic and environmental variation125 and thus improve inference of selection dynamics126,127. A study in Atlantic cod (Gadus morhua) analysed DNA from archived otoliths to search for signatures of divergent selection over a 78-year climatically variable period128. The genetic composition of some populations was temporally stable, yet complete population replacement was evident at others, and increased temporal changes at several loci suggested that adaptation to environmental change had occurred. These findings illustrate the power of spatiotemporal population genomics to inform future conservation efforts.
Molecular phenotypic plasticity in GCC
Phenotypic plasticity is often the main mechanism that allows populations to cope with GCC3,129, with transcriptomics, epigenomics and proteomics being increasingly used to investigate functional plastic responses. Plasticity studies are helpful to understand the range of possible phenotypic responses to environmental change that can influence fitness within a single generation without differential mortality owing to selection. Gene expression23,130,131, epigenetic mechanisms19,132,133 and the proteome134 respond to temperature and environmental changes. Transcription often underlies plastic proteomic and phenotypic changes, whereas epigenetic mechanisms heritably affect transcriptional states in response to the environment135,136. Plastic responses can preclude or delay genetic adaptation if they lead to environmental acclimation137,138. They can also increase phenotypic variation and evolutionary potential129,137,139, which could prove crucial for genetically impoverished populations that experience GCC137, although lack of plasticity or maladaptive plasticity could negatively affect fitness140,141. Organisms can evolve or lose the capacity for transcriptional and epigenetic plasticity, as observed in studies of threespine stickleback (Gasterosteus aculeatus) colonizing freshwater environments142,143 and various populations at range edges144. Plastic and genetic changes can also act synergistically in response to GCC, with species acclimatizing through plastic changes while genetic adaptation ‘catches up’138.
Molecular plasticity studies are shifting towards whole-genome, epigenome, transcriptome and proteome methods owing to decreasing costs. Many studies have assessed transcriptional responses to temperature9,145, with recent studies assessing alternative splicing in thermal acclimation146,147,148,149, which can diversify an organism’s transcriptome. Epigenetic mechanisms provide a mechanistic basis for transcriptional responses to environmental changes associated with GCC. There is a strong focus on differential DNA methylation analysis associated with thermal regime150,151,152, although histone modifications and non-coding RNA expression147,153,154,155 also respond to GCC. Methods such as assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) can directly assess chromatin accessibility, as shown through chromatin conformation and transcriptional changes due to temperature in symbiotic sea anemone (Exaiptasia pallida)156. Proteomics can also be used to assess functional protein responses and resilience to GCC134, which are sometimes driven by transcriptional and epigenomic variation. Although plastic mechanisms clearly respond to temperature and GCC, the integration of both genomic and transcriptomic or epigenomic data is imperative to disentangle genetic from plastic phenotypic effects.
Plasticity and genetics both affect phenotype, although the relative importance of each in the resulting phenotype is unclear. This is further complicated by SNP variation often exerting some control over transcription (for example, expression quantitative trait loci (eQTLs)) and epigenetic state (for example, methylQTLs). For example, seasonal and latitudinal allele frequency changes are associated with thermal response at previously identified eQTLs20 in Drosophila. Both latitudinal and seasonal eQTLs were associated with chromosomal inversion breakpoints21,157, but seasonal eQTLs were not consistent between sites, suggesting that sites may differ in the selective pressures imposed by seasonal changes21. Epigenetic variation and transcription can be linked through methyl-eQTLs, site-specific methylation levels that are maximally correlated with gene expression, which have mostly been used in medical studies158,159 but could be implemented in ecological studies.
The simultaneous assessment of multi-omic data is needed to disentangle the relative importance of genetics versus plasticity for phenotype and to determine the extent of genetic constraint over plasticity. First, studies should assess the extent of genetic control over transcriptional and epigenetic variation, which is present in some systems (for example, Micrarchus stick insects)160 but not others (for example, branching staghorn coral Acropora cervicornis)161. Next, we need to understand how genetic control over plastic mechanisms changes depending on environmental context. For instance, combined transcriptome, methylome and phenotype data revealed genotype by environment effects on growth potential in transgenic Coho salmon (Oncorhynchus kisutch)162. We should further determine under what circumstances organisms evolve differential capacity for plasticity (for example, the aforementioned stickleback studies)142,143 and when genetic variation becomes coupled or uncoupled from transcriptional or epigenomic variation (for example, using whole-genome DNA methylation data and predictive modelling in Arabidopsis thaliana)163.
Finally, multi-omic data must be related to phenotype to reinforce the importance of plasticity in GCC responses. Plasticity studies to measure fitness-related outcomes cement plasticity as an important evolutionary response to GCC9,164 (Fig. 6). Epigenome-wide association studies (EWAS) could be used to link epigenetic and phenotypic changes; although EWAS is primarily used in human disease studies, it was recently used to link methylation changes with temperature fluctuations in humans165 and could thus be used to identify loci involved in thermal response and tolerance. A genomic reaction norm could also be incorporated in molecular plasticity studies to understand the combined effects of environment, genotype, phenotype and demographic characteristics166. The environment is an important consideration in these studies, as phenotype cannot be generalized across multiple environments. These issues can be addressed using common garden experiments or by assessing phenotypic plasticity across many environmental contexts.

When populations exist in different thermal environments, they can develop transcriptional and epigenetic differences, potentially due to thermal acclimation or adaptation. The capacity for plasticity in response to changing thermal environments can affect the persistence of organisms enduring climate change. a, Sandoval-Castillo et al.9 showed that divergent selection on gene expression led to non-parallel transcriptional responses to thermal stress that differed between subtropical, temperate and desert Australian rainbowfish ecotypes (Melanotaenia spp.). RNA sequencing detected 34,815 transcripts among the three ecotypes; 236 genes responded to temperature treatment, with only ten expression changes shared between two ecotypes and five shared between all three. The ecotypes had different thermal tolerances as measured by CTmax, with warm-adapted subtropical rainbowfish showing the greatest tolerance for warming and capacity for transcriptional plasticity. b, Dixon et al.164 reciprocally transplanted stony coral (Acropora millepora) from two different thermal environments and used methyl-binding domain sequencing to analyse gene body DNA methylation of 27,084 genes. Corals that were able to assume the methylation state of local corals had improved fitness-related traits relative to corals that did not resemble local corals epigenetically. Methylation differences were also correlated with transcription, as measured using TagSeq. c, Hypothetical effects of transcriptional and epigenetic plasticity in response to altered thermal environment: (1) if organisms are unable to respond to their environment through transcriptional or epigenetic changes, resulting in an inability to cope with changing temperatures, this could lead to increased mortality; (2) if organisms can adopt the transcriptional state or DNA methylation patterns of local conspecifics, plasticity could lead to increased fitness in altered thermal environments164; and (3) if populations differ in their capacity for plasticity (for example, owing to divergent selection from different thermal environments) or the nature of plastic responses they show (for example, different genes), they may show divergent plastic responses to altered temperature environments. This could lead to populations exhibiting differences in resistance to, and potential persistence in, novel thermal environments9.
eDNA community monitoring and GCC
The analysis of environmental DNA (eDNA) enables the identification of organisms on the basis of their DNA released into the environment (water, soil or air), for example, through skin cells or metabolic waste. This noninvasive genetic approach can predict organism presence and/or absence and sometimes species’ relative abundance167,168. High-throughput sequencing offers the possibility to assess overall biodiversity by simultaneously identifying multiple species through eDNA metabarcoding, which is being used to document community structure in both aquatic and terrestrial ecosystems169. More recently, eDNA metabarcoding protocols have been developed to detect species occurrence in airborne DNA samples170,171,172.
In the context of GCC, eDNA metabarcoding can be used to track shifts in community composition (animals, plants and microorganisms) across time, depending on the environmental selective pressures that occur when the DNA of organisms is trapped in substrate. Two approaches are used to study community composition changes with GCC: laboratory or mesocosm-based experiments173,174 (Fig. 7a), which offer a high degree of control; and time series sampling across a spatial gradient175,176,177 (Fig. 7b), which leverages the high variability offered by the local environment to predict which species will decline or burst relative to those present today174,176. Metagenomics and shotgun sequencing, which are generally more microorganism-focused than metabarcoding, can also inform about temporal variation in community composition (reviewed elsewhere178). A controlled laboratory experiment showed that coral-associated microbiota shifted from a community composition associated with healthy corals to one generally found on diseased corals in abiotic conditions that mimic future GCC179.

a, Ferguson et al.174 used marine mesocosms to examine the impacts of warming, nutrient enrichment and altered top-predator population size structure (common shore crab Carcinus maenas) on coastal microbial biofilm communities in a crossed experimental design. An environmental DNA (eDNA) metabarcoding approach was used by the authors. Based on Illumina MiSeq sequencing of the 16S mitochondrial rRNA gene, they showed that warming and top-predator population size structure both affected bacterial biofilm community composition. Warming increased the abundance of bacteria capable of increased mineralization of dissolved and particulate organic matter, such as flavobacteria, Sphingobacteria and Cytophagia. b. Gallego et al.176 took advantage of the existence of a wide variety of ecosystems within two regions (San Juan Island and the Hood Canal) of the Puget Sound in Washington (USA) to test the effect of environmental parameters on zooplankton marine communities under conditions expected worldwide in the near future. Despite their geographical proximity, these two regions experience substantial differences in environmental conditions (in terms of temperature, pH and CO2), with the Hood Canal resembling future conditions in temperate areas worldwide. The eDNA metabarcoding approach used by the authors included Illumina MiSeq sequencing of the COI mitochondrial gene. They identified distinct communities in warmer and more acidified conditions, with overall reduced richness in diatom assemblages and increased richness in dinoflagellates. The authors used their results to build models to forecast near-term community changes and to determine the probability of the presence of each individual taxon for 2095 compared with 2017. Their results suggest a possible change in relative dominance between diatoms and other phytoplankton species such as dinoflagellates. They also highlight an increased environmental suitability for the coccolithophore Emiliania huxleyi and the harmful Alexandrium sp. mtDNA, mitochondrial DNA. Bottom panel in part a is adapted from ref. 174, CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/). Map and bottom panel in part b adapted with permission from ref. 176, The Royal Society.
At the spatial level, eDNA metabarcoding and metagenomics allow monitoring of biogeographical community range shifts and investigate how animals and plants can cope with changing environmental conditions by migrating to sites that are more favourable to their biology178,180,181. These studies often combine eDNA results with computing projections and machine learning to build habitat occupancy models for species under different GCC scenarios. Metabarcoding data collected during five consecutive years was used to model diatom richness in response to projected GCC and predicted range shifts towards the poles180. Combining genomics with other approaches is particularly helpful for managing invasive species, whose predicted geographical expansion might affect native species ranges during GCC182,183.
In the context of GCC, eDNA metabarcoding can be used to track temporal shifts in community composition associated with environmental change, relying on the DNA of organisms becoming trapped in substrate184. Ancient DNA (aDNA)185,186 can infer previous community composition with greater taxonomic resolution, spatial and temporal precision than other palaeoecological methods184,187. However, its application is more challenging compared with eDNA because of DNA degradation, modification and strand damage that occur with time. Also, sequencing aDNA is challenging as only short reads are generally attainable184. The analysis of aDNA retrieved from permafrost and sediments can allow documentation of community composition up to 50,000 years ago185,187. The oldest aDNA discovered to date was 2-million-year-old DNA trapped in North Greenland ice sediment, which was linked to a rich assemblage of plants and animals188. aDNA analysis offers fine resolution to reconstruct temporal dynamics of community shifts184,187,189,190,191,192. aDNA extracted from fossil rodent middens, small piles of seeds, bones or leaves gathered by rodents, allowed reconstruction of late Quaternary vegetation dynamics, showing perennial species displacement ~1,000 m downhill during pluvial events191. aDNA analysis can also track extirpations and extinctions185 associated with glacial events in mammals193 and plants194. Reconstruction of community responses to past environmental events provides a unique opportunity to forecast how contemporary communities will change with GCC. Extinction models based on aDNA data that link community fluctuations with environmental variation can elucidate the role of humans in these processes195.
eDNA, aDNA metabarcoding and metagenomics provide new insights into community responses to GCC, but do not predict whether new species will colonize an environment or how species adapt to future conditions. Neither do they differentiate between living and dead organisms185. Environmental RNA (eRNA) offers a promising avenue of research that could pave the way to new disciplines, including environmental transcriptomics, which could be used to noninvasively assess at the community level the role of plasticity in coping with GCC196. However, rapid medical, forensic and environmental advances in eDNA analysis are raising privacy and ethical concerns, requiring future legal considerations197.
The above approaches are not mutually exclusive. Holistic community reconstructions will require the integration of various methodologies such as the use of multiple markers in metabarcoding, both universal and more specific primers, and shotgun sequencing. This will ultimately allow a better understanding of how a broad array of taxa will react to GCC, which is crucial to understand, as heterogeneous taxonomic groups may show contrasting responses to GCC198.
Conclusions and perspectives
Our capacity to monitor and understand species’ responses to GCC will be enhanced by integration of multi-omics methods with other types of data5,6,60. Studies considering several evolutionary processes to predict the fate of species are steps towards a more comprehensive integration of population genomics with GCC science199,200,201,202. Moreover, multi-omic data sets will enhance studies on the evolution of plasticity and its relationship to genomic variation, furthering our understanding of how and when plasticity can aid species experiencing GCC. Plasticity has commonly been considered separate from adaptive variation, which is often assumed to be genetic, although recent studies showing plastic contributions to fitness and adaptation or maladaptation suggest that plasticity should be considered in tandem with other sources of adaptive variation. Future incorporation of other genomic methods into plasticity studies (for example, landscape transcriptomics, epigenomics and proteomics; GEA-type analyses) would further improve our understanding of the role of plasticity in GCC response and of when plasticity and genetic adaptation occur independently or in tandem.
Relating epigenomic and genomic variation to phenotype and fitness in different environments through time could improve our understanding of species’ evolutionary potential in the face of GCC203. Although this is still challenging, it is becoming feasible with methodological advances that assess multi-omics contributions to phenotypes204,205,206,207 and their underlying genetic architecture204. Such an integrative approach could provide greater insight into the molecular basis of complex phenotypes that confer environmental adaptation compared with the analysis of a single type of omic data set. The rise of long-read sequencing has largely enabled the discovery of structural variation, revealing the contribution of large-effect mutations in genetic adaptation208,209,210,211. However, like single SNPs, the functional and the fitness consequences of their polymorphism remain mostly unknown. Integrating multi-omics methods with resurrection ecology or E&R experiments would be particularly powerful. Epigenomic or genomic variation between ancestors and descendants can also be compared to determine the molecular basis of phenotypic change through generations212. Once ecologically relevant genes are identified, their phenotypic effects and genetic architecture can be confirmed through methods that include controlled breeding designs and CRISPR editing113. However, some epigenetic or genetic variation may show no association with phenotype or fitness, or may show variable fitness outcomes in different environments, which may limit our ability to relate multi-omic variation to tangible GCC responses.
Advances in biomarker detection techniques could improve ecosystem monitoring and assessment of adaptive potential during GCC. Liquid biopsy could be used to noninvasively develop multi-omic biomarkers for ecosystem health, including microbial community composition213. Epigenetic biomarkers can provide information on past thermal stress214 and demographic characteristics (for example, sex214 or age215) and could be used to identify biomarkers of thermal resilience in natural populations, although there are still few studies that have analysed epigenetic biomarkers in the context of GCC.
Modelling techniques can predict species’ responses to environmental change through time and provide an efficient means to relate temporal multi-omic variation to GCC-induced phenotypic and fitness consequences. Machine learning can aid in biomarker detection214 and predict future consequences of GCC for organisms through forward modelling, whereby predictions are made about outcomes or the behaviour of a system given a particular model216. Improvements to individual-based modelling (IBM) frameworks provide a powerful way to explore impacts of GCC on species’ persistence under increasingly complex eco-evolutionary scenarios217. Integral projection models are also used to model demographic changes due to genetic variation218 and could be expanded to use large-scale genomic data in the context of GCC. Integrating eDNA metabarcoding and modelling can aid in inferring the effects of environmental, geographical and socioeconomic factors on community diversity219. However, biomarkers and novel modelling approaches will require critical evaluation to assess their utility in predicting GCC responses in natural systems.
Genomics methods have provided insights into how GCC affects species, although increased availability of genomic resources and open data are needed. Reference genomes are becoming increasingly available through initiatives such as the Earth Biogenome Project220, thus improving the feasibility of studies in non-model organisms, although many remain understudied. For example, primary producers such as microbial eukaryotes are crucial for ecosystem function, although the effects of GCC on microbes, their genomes and subsequent ecosystem effects are unclear. Requirements for open data and reproducible code221, standardized data reporting and availability of georeferenced environmental data222 will facilitate meta-analyses223 and conservation macrogenetics224,225. Successful conservation and monitoring during GCC will require the development of science–policy–society interactions226,227 and continual reassessment of evolving omics approaches to inform decision-making. Holistic applications of omics tools could mitigate the effects of GCC on biodiversity, contribute to restoring ecosystems and improve health, food security and a sustainable global bioeconomy.
References
-
Smith, T. B. & Bernatchez, L. Evolutionary change in human-altered environments. Mol. Ecol. 17, 1–8 (2008).
-
Bellard, C., Bertelsmeier, C., Leadley, P., Thuiller, W. & Courchamp, F. Impacts of climate change on the future of biodiversity. Ecol. Lett. 15, 365–377 (2012).
-
Merilä, J. & Hendry, A. P. Climate change, adaptation, and phenotypic plasticity: the problem and the evidence. Evol. Appl. 7, 1–14 (2014).
-
Scheffers, B. R. et al. The broad footprint of climate change from genes to biomes to people. Science 354, aaf7671 (2016).
-
Waldvogel, A.-M. et al. Evolutionary genomics can improve prediction of species’ responses to climate change. Evol. Lett. 4, 4–18 (2020). A road map of how science and society can work together to facilitate sampling, estimating of fitness parameters and genome sequencing for a broad range of species to implement mitigation measures to face GCC.
-
Lancaster, L. T. et al. Understanding climate change response in the age of genomics. J. Anim. Ecol. 91, 1056–1063 (2022). Special issue highlighting how emerging genomic approaches are used to understand population responses to GCC across a diverse range of animal systems.
-
Huey, R. B. et al. Predicting organismal vulnerability to climate warming: roles of behaviour, physiology and adaptation. Phil. Trans. R. Soc. B 367, 1665–1679 (2012).
-
Klein, S. G. et al. Projecting coral responses to intensifying marine heatwaves under ocean acidification. Glob. Change Biol. 28, 1753–1765 (2022).
-
Sandoval-Castillo, J. et al. Adaptation of plasticity to projected maximum temperatures and across climatically defined bioregions. Proc. Natl Acad. Sci. USA 117, 17112–17121 (2020).
-
Eisenhauer, N. et al. The dark side of animal phenology. Trends Ecol. Evol. 33, 898–901 (2018).
-
Miller-Rushing, A. J. & Primack, R. B. Global warming and flowering times in Thoreau’s Concord: a community perspective. Ecology 89, 332–341 (2008).
-
Bruno, J. F. et al. Thermal stress and coral cover as drivers of coral disease outbreaks. PLOS Biol. 5, e124 (2007).
-
Gilman, S. E., Urban, M. C., Tewksbury, J., Gilchrist, G. W. & Holt, R. D. A framework for community interactions under climate change. Trends Ecol. Evol. 25, 325–331 (2010).
-
Parmesan, C. & Singer, M. C. Mosaics of climatic stress across species’ ranges: tradeoffs cause adaptive evolution to limits of climatic tolerance. Phil. Trans. R. Soc. B 377, 20210003 (2022).
-
Hoffmann, A. A. & Sgrò, C. M. Climate change and evolutionary adaptation. Nature 470, 479–485 (2011).
-
Carroll, S. P. et al. Applying evolutionary biology to address global challenges. Science 346, 1245993 (2014).
-
Donelson, J. M. et al. Understanding interactions between plasticity, adaptation and range shifts in response to marine environmental change. Phil. Trans. R. Soc. B 374, 20180186 (2019).
-
Hansen, M. M., Olivieri, I., Waller, D. M., Nielsen, E. E. & Group, T. G. W. Monitoring adaptive genetic responses to environmental change. Mol. Ecol. 21, 1311–1329 (2012).
-
Verhoeven, K. J. F., vonHoldt, B. M. & Sork, V. L. Epigenetics in ecology and evolution: what we know and what we need to know. Mol. Ecol. 25, 1631–1638 (2016).
-
Everett, L. J. et al. Gene expression networks in the Drosophila genetic reference panel. Genome Res. 30, 485–496 (2020).
-
Yu, Y. & Bergland, A. O. Distinct signals of clinal and seasonal allele frequency change at eQTLs in Drosophila melanogaster. Evol 76, 2758–2768 (2022).
-
Stange, M., Barrett, R. D. H. & Hendry, A. P. The importance of genomic variation for biodiversity, ecosystems and people. Nat. Rev. Genet. 22, 89–105 (2021).
-
McGaughran, A., Laver, R. & Fraser, C. Evolutionary responses to warming. Trends Ecol. Evol. 36, 591–600 (2021).
-
Springer, N. M. & Schmitz, R. J. Exploiting induced and natural epigenetic variation for crop improvement. Nat. Rev. Genet. 18, 563–575 (2017).
-
Logsdon, G. A., Vollger, M. R. & Eichler, E. E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 21, 597–614 (2020).
-
De Coster, W., Weissensteiner, M. H. & Sedlazeck, F. J. Towards population-scale long-read sequencing. Nat. Rev. Genet. 22, 572–587 (2021).
-
Thomas, L. et al. Spatially varying selection between habitats drives physiological shifts and local adaptation in a broadcast spawning coral on a remote atoll in Western Australia. Sci. Adv. 8, eabl9185 (2022).
-
Hoban, S. et al. Finding the genomic basis of local adaptation: pitfalls, practical solutions, and future directions. Am. Nat. 188, 379–397 (2016).
-
Boulanger, E. et al. Climate differently influences the genomic patterns of two sympatric marine fish species. J. Anim. Ecol. 91, 1180–1195 (2022).
-
Rellstab, C., Gugerli, F., Eckert, A. J., Hancock, A. M. & Holderegger, R. A practical guide to environmental association analysis in landscape genomics. Mol. Ecol. 24, 4348–4370 (2015).
-
Lasky, J. R., Josephs, E. B. & Morris, G. P. Genotype–environment associations to reveal the molecular basis of environmental adaptation. Plant Cell 35, 125–138 (2023).
-
Alvarado, A. H. et al. Genotype–environment associations across spatial scales reveal the importance of putative adaptive genetic variation in divergence. Evol. Appl. 15, 1390–1407 (2022).
-
Nielsen, E. S., Henriques, R., Beger, M., Toonen, R. J. & Von der Heyden, S. Multi-model seascape genomics identifies distinct environmental drivers of selection among sympatric marine species. BMC Evol. Biol. 20, 1–17 (2020).
-
Brauer, C. J., Unmack, P. J., Smith, S., Bernatchez, L. & Beheregaray, L. B. On the roles of landscape heterogeneity and environmental variation in determining population genomic structure in a dendritic system. Mol. Ecol. 27, 3484–3497 (2018).
-
Grummer, J. A. et al. Aquatic landscape genomics and environmental effects on genetic variation. Trends Ecol. Evol. 34, 641–654 (2019).
-
Lotterhos, K. E. & Whitlock, M. C. The relative power of genome scans to detect local adaptation depends on sampling design and statistical method. Mol. Ecol. 24, 1031–1046 (2015).
-
Forester, B. R., Lasky, J. R., Wagner, H. H. & Urban, D. L. Comparing methods for detecting multilocus adaptation with multivariate genotype–environment associations. Mol. Ecol. 27, 2215–2233 (2018).
-
Capblancq, T., Luu, K., Blum, M. G. & Bazin, E. Evaluation of redundancy analysis to identify signatures of local adaptation. Mol. Ecol. Resour. 18, 1223–1233 (2018).
-
Martínez-Berdeja, A. et al. Functional variants of DOG1 control seed chilling responses and variation in seasonal life-history strategies in Arabidopsis thaliana. Proc. Natl Acad. Sci. USA 117, 2526–2534 (2020).
-
Fournier‐Level, A. et al. Adaptive significance of flowering time variation across natural seasonal environments in Arabidopsis thaliana. N. Phytol. 234, 719–734 (2022).
-
Capblancq, T. & Forester, B. R. Redundancy analysis: a Swiss Army knife for landscape genomics. Methods Ecol. Evol. 12, 2298–2309 (2021). A review of the application and challenges of RDA for understanding the relationship between genetic variation and the environment, with a case study and associated tutorial for users.
-
Booker, T. R., Yeaman, S., Whiting, J. R. & Whitlock, M. C. The WZA: a window-based method for characterizing genotype-environment associations. Mol. Ecol. Resour. https://doi.org/10.1111/1755-0998.13768 (2023).
-
Meek, M. H. et al. Understanding local adaptation to prepare populations for climate change. Bioscience 73, 36–47 (2023).
-
Beer, M. A., Kane, R. A., Micheletti, S. J., Kozakiewicz, C. P. & Storfer, A. Landscape genomics of the streamside salamander: implications for species management in the face of environmental change. Evol. Appl. 15, 220–236 (2022). A demonstration of the use of GEAs to detect adaptive variation for understanding the potential for adaptation to environmental challenges across a heterogeneous landscape.
-
Aguirre‐Liguori, J. et al. Connecting genomic patterns of local adaptation and niche suitability in teosintes. Mol. Ecol. 26, 4226–4240 (2017).
-
Flanagan, S. P., Forester, B. R., Latch, E. K., Aitken, S. N. & Hoban, S. Guidelines for planning genomic assessment and monitoring of locally adaptive variation to inform species conservation. Evol. Appl. 11, 1035–1052 (2018).
-
Xuereb, A., d’Aloia, C. C., Andrello, M., Bernatchez, L. & Fortin, M. J. Incorporating putatively neutral and adaptive genomic data into marine conservation planning. Conserv. Biol. 35, 909–920 (2020).
-
Forester, B. R. et al. Genomics‐informed delineation of conservation units in a desert amphibian. Mol. Ecol. 31, 5249–5269 (2022).
-
Mahony, C. R. et al. Evaluating genomic data for management of local adaptation in a changing climate: a lodgepole pine case study. Evol. Appl. 13, 116–131 (2020).
-
Chen, Z. et al. Applying genomics in assisted migration under climate change: framework, empirical applications, and case studies. Evol. Appl. 15, 3–21 (2021).
-
Lotterhos, K. E. The paradox of adaptive trait clines with nonclinal patterns in the underlying genes. Proc. Natl Acad. Sci. USA 120, e2220313120 (2023).
-
Rockman, M. V. THE QTN program and the alleles that matter for evolution: all that’s gold does not glitter. Evolution 66, 1–17 (2012).
-
Rougemont, Q. et al. Long-distance migration is a major factor driving local adaptation at continental scale in Coho salmon. Mol. Ecol. 32, 542–559 (2023).
-
Rybnikov, S. R., Frenkel, Z., Hübner, S., Weissman, D. B. & Korol, A. B. Modeling the evolution of recombination plasticity: a prospective review. BioEssays 45, e2200237 (2023).
-
Schlötterer, C. How predictable is adaptation from standing genetic variation? Experimental evolution in Drosophila highlights the central role of redundancy and linkage disequilibrium. Phil. Trans. R. Soc. B 378, 20220046 (2023).
-
Exposito-Alonso, M., Burbano, H. A., Bossdorf, O., Nielsen, R. & Weigel, D. Natural selection on the Arabidopsis thaliana genome in present and future climates. Nature 573, 126–129 (2019). A large-scale common garden experiment showing differences in relative fitness under climate change associated with candidate genomic regions in Arabidopsis thaliana.
-
Mitchell-Olds, T. & Schmitt, J. Genetic mechanisms and evolutionary significance of natural variation in Arabidopsis. Nature 441, 947–952 (2006).
-
Fitzpatrick, M. C. & Keller, S. R. Ecological genomics meets community-level modelling of biodiversity: mapping the genomic landscape of current and future environmental adaptation. Ecol. Lett. 18, 1–16 (2015). Adapted the GF approach on SNP data set, originally developed to model spatial variation in community composition, to model turnover in allele frequency and called it ‘genetic offset’.
-
Bay, R. A. et al. Genomic signals of selection predict climate-driven population declines in a migratory bird. Science 359, 83–86 (2018). Among the first studies to assess genomic offset on natural populations and link it with observed losses in fitness (population declines) in order to provide a rigorous validation of the predictions.
-
Capblancq, T., Fitzpatrick, M. C., Bay, R. A., Exposito-Alonso, M. & Keller, S. R. Genomic prediction of (mal)adaptation across current and future climatic landscapes. Annu. Rev. Ecol. Evol. Syst. 51, 245–269 (2020). Review of the main steps and associated statistical methods in genomic prediction of maladaptation across current and future climatic landscapes.
-
Rellstab, C., Dauphin, B. & Exposito-Alonso, M. Prospects and limitations of genomic offset in conservation management. Evol. Appl. 14, 1202–1212 (2021).
-
Ellis, N., Smith, S. J. & Pitcher, C. R. Gradient forests: calculating importance gradients on physical predictors. Ecology 93, 156–168 (2012).
-
Ingvarsson, P. K. & Bernhardsson, C. Genome-wide signatures of environmental adaptation in European aspen (Populus tremula) under current and future climate conditions. Evol. Appl. 13, 132–142 (2019).
-
Martins, K. et al. Landscape genomics provides evidence of climate-associated genetic variation in Mexican populations of Quercus rugosa. Evol. Appl. 11, 1842–1858 (2018).
-
Ruegg, K. et al. Ecological genomics predicts climate vulnerability in an endangered southwestern songbird. Ecol. Lett. 21, 1085–1096 (2018).
-
Ferrier, S. & Guisan, A. Spatial modelling of biodiversity at the community level. J. Appl. Ecol. 43, 393–404 (2006).
-
Supple, M. A. et al. Landscape genomic prediction for restoration of a Eucalyptus foundation species under climate change. eLife 7, e31835 (2018).
-
Steane, D. A. et al. Genome-wide scans detect adaptation to aridity in a widespread forest tree species. Mol. Ecol. 23, 2500–2513 (2014).
-
Carvalho, C. S. et al. Combining genotype, phenotype, and environmental data to delineate site-adjusted provenance strategies for ecological restoration. Mol. Ecol. Resour. 21, 44–58 (2021).
-
Rellstab, C. et al. Signatures of local adaptation in candidate genes of oaks (Quercus spp.) with respect to present and future climatic conditions. Mol. Ecol. 25, 5907–5924 (2016).
-
Pina-Martins, F., Baptista, J., Pappas, G. Jr & Paulo, O. S. New insights into adaptation and population structure of cork oak using genotyping by sequencing. Glob. Change Biol. 25, 337–350 (2019).
-
Rochat, E., Selmoni, O. & Joost, S. Spatial areas of genotype probability: predicting the spatial distribution of adaptive genetic variants under future climatic conditions. Divers. Distrib. 27, 1076–1090 (2021).
-
Gain, C. et al. A quantitative theory for genomic offset statistics. Mol. Biol. Evol. 40, 6 (2023).
-
Hoffmann, A. A., Weeks, A. R. & Sgrò, C. M. Opportunities and challenges in assessing climate change vulnerability through genomics. Cell 184, 1420–1425 (2021). Describes the limitations and their respective solutions in genomic vulnerability assessments.
-
Aguirre-Liguori, J. A., Ramírez-Barahona, S. & Gaut, B. S. The evolutionary genomics of species’ responses to climate change. Nat. Ecol. Evol. 5, 1350–1360 (2021).
-
Aguirre‐Liguori, J. A. et al. Divergence with gene flow is driven by local adaptation to temperature and soil phosphorus concentration in teosinte subspecies (Zea mays parviglumis and Zea mays mexicana). Mol. Ecol. 28, 2814–2830 (2019).
-
Brauer, C. J. et al. Natural hybridization reduces vulnerability to climate change. Nat. Clim. Change 13, 282–289 (2023).
-
Rhoné, B. et al. Pearl millet genomic vulnerability to climate change in West Africa highlights the need for regional collaboration. Nat. Commun. 11, 5274 (2020).
-
Weider, L. J., Jeyasingh, P. D. & Frisch, D. Evolutionary aspects of resurrection ecology: progress, scope, and applications — an overview. Evol. Appl. 11, 3–10 (2017).
-
Kawecki, T. J. et al. Experimental evolution. Trends Ecol. Evol. 27, 547–560 (2012). Excellent review paper on main strengths and weaknesses of experimental evolution.
-
Kofler, R. & Schlötterer, C. A guide for the design of evolve and resequencing studies. Mol. Biol. Evol. 31, 474–483 (2014). A paper that used simulations to propose guidelines for optimizing design of E&R studies.
-
Lenski, R. E., Rose, M. R., Simpson, S. C. & Tadler, S. C. Long-term experimental evolution in Escherichia coli. I. Adaptation and divergence during 2,000 generations. Am. Nat. 138, 1315–1341 (1991).
-
Elena, S. F. & Lenski, R. E. Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat. Rev. Genet. 4, 457–469 (2003). A landmark review paper establishing E&R experiments as a new field of research developed around the idea of using microorganisms to investigate the dynamics of evolutionary adaptation.
-
Barrick, J. E. et al. Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461, 1243–1247 (2009).
-
Tenaillon, O. et al. Tempo and mode of genome evolution in a 50,000-generation experiment. Nature 536, 165–170 (2016).
-
Good, B. H., McDonald, M. J., Barrick, J. E., Lenski, R. E. & Desai, M. M. The dynamics of molecular evolution over 60,000 generations. Nature 551, 45–50 (2017).
-
Lenski, R. E. Revisiting the design of the long-term evolution experiment with Escherichia coli. J. Mol. Evol. 91, 241–253 (2023).
-
Barghi, N. et al. Genetic redundancy fuels polygenic adaptation in Drosophila. PLoS Biol. 17, e3000128 (2019).
-
Long, A., Liti, G., Luptak, A. & Tenaillon, O. Elucidating the molecular architecture of adaptation via evolve and resequence experiments. Nat. Rev. Genet. 16, 567–582 (2015). A comprehensive paper that reviews the field of E&R experiments across diverse systems, ranging from simple non-living RNA to bacteria, yeast and Drosophila sp.
-
Schlötterer, C., Kofler, R., Versace, E., Tobler, R. & Franssen, S. Combining experimental evolution with next-generation sequencing: a powerful tool to study adaptation from standing genetic variation. Heredity 114, 431–440 (2015).
-
Schlötterer, C., Tobler, R., Kofler, R. & Nolte, V. Sequencing pools of individuals—mining genome-wide polymorphism data without big funding. Nat. Rev. Genet. 15, 749–763 (2014).
-
Huang, C.-J., Lu, M.-Y., Chang, Y.-W. & Li, W.-H. Experimental evolution of yeast for high-temperature tolerance. Mol. Biol. Evol. 35, 1823–1839 (2018).
-
Otte, K. A., Nolte, V., Mallard, F. & Schlötterer, C. The genetic architecture of temperature adaptation is shaped by population ancestry and not by selection regime. Genome Biol. 22, 211 (2021).
-
Burny, C., Nolte, V., Dolezal, M. & Schlötterer, C. Genome-wide selection signatures reveal widespread synergistic effects of two different stressors in Drosophila melanogaster. Proc. Biol. Sci. 289, 20221857 (2022).
-
Orozco‐Terwengel, P. et al. Adaptation of Drosophila to a novel laboratory environment reveals temporally heterogeneous trajectories of selected alleles. Mol. Ecol. 21, 4931–4941 (2012).
-
Tobler, R. et al. Massive habitat-specific genomic response in D. melanogaster populations during experimental evolution in hot and cold environments. Mol. Biol. Evol. 31, 364–375 (2014).
-
Brennan, R. S., Garrett, A. D., Huber, K. E., Hargarten, H. & Pespeni, M. H. Rare genetic variation and balanced polymorphisms are important for survival in global change conditions. Proc. R. Soc. B. 286, 20190943 (2019).
-
Pespeni, M. H. et al. Evolutionary change during experimental ocean acidification. Proc. Natl Acad. Sci. USA 110, 6937–6942 (2013).
-
Waldvogel, A. M. et al. The genomic footprint of climate adaptation in Chironomus riparius. Mol. Ecol. 27, 1439–1456 (2018).
-
Mérot, C., Llaurens, V., Normandeau, E., Bernatchez, L. & Wellenreuther, M. Balancing selection via life-history trade-offs maintains an inversion polymorphism in a seaweed fly. Nat. Commun. 11, 670 (2020).
-
Hsu, S. K., Belmouaden, C., Nolte, V. & Schlötterer, C. Parallel gene expression evolution in natural and laboratory evolved populations. Mol. Ecol. 30, 884–894 (2021).
-
Pfenninger, M. & Foucault, Q. Genomic processes underlying rapid adaptation of a natural Chironomus riparius population to unintendedly applied experimental selection pressures. Mol. Ecol. 29, 536–548 (2020).
-
Orsini, L. et al. The evolutionary time machine: using dormant propagules to forecast how populations can adapt to changing environments. Trends Ecol. Evol. 28, 274–282 (2013). An excellent, early review on how combining resurrection ecology and genomics can enhance capacity to forecast how populations can adapt to changing environments.
-
Kerfoot, W. C., Robbins, J. A. & Weider, L. J. A new approach to historical reconstruction: combining descriptive and experimental paleolimnology. Limnol. Oceanogr. 44, 1232–1247 (1999).
-
Kerfoot, W. C. & Weider, L. J. Experimental paleoecology (resurrection ecology): chasing Van Valen’s Red Queen hypothesis. Limnol. Oceanogr. 49, 1300–1316 (2004).
-
Franks, S. J. et al. The resurrection initiative: storing ancestral genotypes to capture evolution in action. Bioscience 58, 870–873 (2008).
-
Franks, S. J., Kane, N. C., O’Hara, N. B., Tittes, S. & Rest, J. S. Rapid genome‐wide evolution in Brassica rapa populations following drought revealed by sequencing of ancestral and descendant gene pools. Mol. Ecol. 25, 3622–3631 (2016).
-
Franks, S. J. & Hoffmann, A. A. Genetics of climate change adaptation. Annu. Rev. Genet. 46, 185–208 (2012). This early, comprehensive review paper covers in detail integrative approaches towards elucidating the genetic basis of adaptation.
-
Orsini, L., Spanier, K. I. & De Meester, L. Genomic signature of natural and anthropogenic stress in wild populations of the waterflea Daphnia magna: validation in space, time and experimental evolution. Mol. Ecol. 21, 2160–2175 (2012).
-
Orsini, L. et al. Temporal genetic stability in natural populations of the waterflea Daphnia magna in response to strong selection pressure. Mol. Ecol. 25, 6024–6038 (2016).
-
Franks, S. J., Hamann, E. & Weis, A. E. Using the resurrection approach to understand contemporary evolution in changing environments. Evol. Appl. 11, 17–28 (2018).
-
Chaturvedi, A. et al. Extensive standing genetic variation from a small number of founders enables rapid adaptation in Daphnia. Nat. Commun. 12, 4306 (2021).
-
Wersebe, M. J. & Weider, L. J. Resurrection genomics provides molecular and phenotypic evidence of rapid adaptation to salinization in a keystone aquatic species. Proc. Natl Acad. Sci. USA 120, e2217276120 (2023).
-
Cuenca Cambronero, M., Zeis, B. & Orsini, L. Haemoglobin‐mediated response to hyper‐thermal stress in the keystone species Daphnia magna. Evol. Appl. 11, 112–120 (2018).
-
Exposito-Alonso, M. et al. The rate and potential relevance of new mutations in a colonizing plant lineage. PLoS Genet. 14, e1007155 (2018).
-
Hamann, E. et al. Plant eco-evolutionary responses to climate change: emerging directions. Plant Sci. 304, 110737 (2021).
-
Frisch, D. et al. A millennial-scale chronicle of evolutionary responses to cultural eutrophication in Daphnia. Ecol. Lett. 17, 360–368 (2014).
-
Hamann, E. et al. Rapid evolutionary changes in gene expression in response to climate fluctuations. Mol. Ecol. 30, 193–206 (2021).
-
Ghalambor, C. K. et al. Non-adaptive plasticity potentiates rapid adaptive evolution of gene expression in nature. Nature 525, 372–375 (2015).
-
Campbell-Staton, S. C. et al. Winter storms drive rapid phenotypic, regulatory, and genomic shifts in the green anole lizard. Science 357, 495–498 (2017).
-
Jensen, E. L. & Leigh, D. M. Using temporal genomics to understand contemporary climate change responses in wildlife. Ecol. Evol. 12, e9340 (2022).
-
Clark, R. D. et al. The practice and promise of temporal genomics for measuring evolutionary responses to global change. Mol. Ecol. Resour. https://doi.org/10.1111/1755-0998.13789 (2023).
-
Elleouet, J. S. & Aitken, S. N. The interplay between demography and neutral evolution at the expansion front of a widespread conifer, Picea sitchensis. Preprint at bioRxiv https://doi.org/10.1101/327742 (2018).
-
Lang, P. L. M., Willems, F. M., Scheepens, J. F., Burbano, H. A. & Bossdorf, O. Using herbaria to study global environmental change. Nat. Phytol. 221, 110–122 (2019).
-
Czorlich, Y., Aykanat, T., Erkinaro, J., Orell, P. & Primmer, C. R. Rapid evolution in salmon life history induced by direct and indirect effects of fishing. Science 376, 420–423 (2022).
-
Buffalo, V. & Coop, G. The linked selection signature of rapid adaptation in temporal genomic data. Genetics 213, 1007–1045 (2019).
-
Foll, M., Shim, H. & Jensen, J. D. WFABC: a Wright–Fisher ABC-based approach for inferring effective population sizes and selection coefficients from time-sampled data. Mol. Ecol. Resour. 15, 87–98 (2015).
-
Therkildsen, N. O. et al. Spatiotemporal SNP analysis reveals pronounced biocomplexity at the northern range margin of Atlantic cod Gadus morhua. Evol. Appl. 6, 690–705 (2013).
-
Anderson, J. T., Panetta, A. M. & Mitchell-Olds, T. Evolutionary and ecological responses to anthropogenic climate change: update on anthropogenic climate change. Plant Physiol. 160, 1728–1740 (2012).
-
DeBiasse, M. B. & Kelly, M. W. Plastic and evolved responses to global change: what can we learn from comparative transcriptomics? J. Hered. 107, 71–81 (2016).
-
Oomen, R. A. & Hutchings, J. A. Transcriptomic responses to environmental change in fishes: insights from RNA sequencing. Facets 2, 610–641 (2017).
-
Hu, J. & Barrett, R. Epigenetics in natural animal populations. J. Evol. Biol. 30, 1612–1632 (2017).
-
McCaw, B. A., Stevenson, T. J. & Lancaster, L. T. Epigenetic responses to temperature and climate. Integr. Comp. Biol. 60, 1469–1480 (2020).
-
Abdelnour, S. A. et al. Stress biomarkers and proteomics alteration to thermal stress in ruminants: a review. J. Therm. Biol. 79, 120–134 (2019).
-
Anastasiadi, D., Venney, C. J., Bernatchez, L. & Wellenreuther, M. Epigenetic inheritance and reproductive mode in plants and animals. Trends Ecol. Evol. 36, 1124–1140 (2021).
-
Ecker, S., Pancaldi, V., Valencia, A., Beck, S. & Paul, D. S. Epigenetic and transcriptional variability shape phenotypic plasticity. BioEssays 40, 1700148 (2018).
-
O’Dea, R. E., Noble, D. W., Johnson, S. L., Hesselson, D. & Nakagawa, S. The role of non-genetic inheritance in evolutionary rescue: epigenetic buffering, heritable bet hedging and epigenetic traps. Environ. Epigenet. 2, dvv014 (2016).
-
Pottier, P. et al. Developmental plasticity in thermal tolerance: ontogenetic variation, persistence, and future directions. Ecol. Lett. 25, 2245–2268 (2022).
-
Gianella, M., Bradford, K. J. & Guzzon, F. Ecological, (epi) genetic and physiological aspects of bet-hedging in angiosperms. Plant Reprod. 34, 21–36 (2021).
-
Donelan, S. C. et al. Transgenerational plasticity in human-altered environments. Trends Ecol. Evol. 35, 115–124 (2020).
-
Morris, M. R. & Rogers, S. M. Overcoming maladaptive plasticity through plastic compensation. Curr. Zool. 59, 526–536 (2013).
-
Hu, J. & Barrett, R. D. The role of plastic and evolved DNA methylation in parallel adaptation of threespine stickleback (Gasterosteus aculeatus). Mol. Ecol. 32, 1581–1591 (2022).
-
Morris, M. R. et al. Gene expression plasticity evolves in response to colonization of freshwater lakes in threespine stickleback. Mol. Ecol. 23, 3226–3240 (2014).
-
Usui, T. et al. The evolution of plasticity at geographic range edges. Trends Ecol. Evol. 38, 831–842 (2023).
-
Bay, R. A. & Palumbi, S. R. Rapid acclimation ability mediated by transcriptome changes in reef-building corals. Genome Biol. Evol. 7, 1602–1612 (2015).
-
Healy, T. M. & Schulte, P. M. Patterns of alternative splicing in response to cold acclimation in fish. J. Exp. Biol. 222, jeb.193516 (2019).
-
Pajoro, A., Severing, E., Angenent, G. & Immink, R. Histone H3 lysine 36 methylation affects temperature-induced alternative splicing and flowering in plants. Genome Biol. 18, 102 (2017).
-
Seo, P. J., Park, M.-J. & Park, C.-M. Alternative splicing of transcription factors in plant responses to low temperature stress: mechanisms and functions. Planta 237, 1415–1424 (2013).
-
Thorstensen, M. J., Turko, A. J., Heath, D. D., Jeffries, K. M. & Pitcher, T. E. Acute thermal stress elicits interactions between gene expression and alternative splicing in a fish of conservation concern. J. Exp. Biol. 225, jeb244162 (2022). Differential gene expression and alternative splicing are assessed owing to handling and thermal stress in the imperiled redside dace (Clinostomus elongatus), with important implications for conservation and reintroductions during GCC.
-
Strader, M., Wong, J., Kozal, L., Leach, T. & Hofmann, G. Parental environments alter DNA methylation in offspring of the purple sea urchin, Strongylocentrotus purpuratus. J. Exp. Mar. Biol. Ecol. 517, 54–64 (2019). A controlled epigenetic inheritance experiment showing that parental exposure to oceanic upwelling, a result of GCC, led to altered offspring DNA methylation associated with body size regardless of offspring environment.
-
Venney, C. J. et al. Thermal regime during parental sexual maturation, but not during offspring rearing, modulates DNA methylation in brook charr (Salvelinus fontinalis). Proc. Biol. Sci. 289, 20220670 (2022).
-
Gugger, P. F., Fitz‐Gibbon, S., PellEgrini, M. & Sork, V. L. Species‐wide patterns of DNA methylation variation in Quercus lobata and their association with climate gradients. Mol. Ecol. 25, 1665–1680 (2016).
-
Ishihara, A., Sapon, M. A. & Yamauchi, K. Seasonal acclimatization and thermal acclimation induce global histone epigenetic changes in liver of bullfrog (Lithobates catesbeianus) tadpole. Comp. Biochem. Physiol. A 230, 39–48 (2019).
-
Hajyzadeh, M., Turktas, M., Khawar, K. M. & Unver, T. miR408 overexpression causes increased drought tolerance in chickpea. Gene 555, 186–193 (2015).
-
Liu, Q. et al. Integrating small RNA sequencing with QTL mapping for identification of miRNAs and their target genes associated with heat tolerance at the flowering stage in rice. Front. Plant Sci. 8, 43 (2017).
-
Weizman, E. & Levy, O. The role of chromatin dynamics under global warming response in the symbiotic coral model Aiptasia. Commun. Biol. 2, 282 (2019). Correlations between RNA expression and promoter chromatin accessibility were found in symbiotic sea anemones exposed to thermal stress in genomic regions associated with oxidative stress and immune response.
-
Machado, H. E. et al. Broad geographic sampling reveals the shared basis and environmental correlates of seasonal adaptation in Drosophila. eLife 10, e67577 (2021).
-
Liu, Y. et al. Methylation-eQTL analysis in cancer research. Bioinformatics 37, 4014–4022 (2021).
-
Yang, I. V. et al. Relationship of DNA methylation and gene expression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 190, 1263–1272 (2014).
-
Dunning, L. T., Dennis, A. B., Sinclair, B. J., Newcomb, R. D. & Buckley, T. R. Divergent transcriptional responses to low temperature among populations of alpine and lowland species of New Zealand stick insects (Micrarchus). Mol. Ecol. 23, 2712–2726 (2014).
-
Parkinson, J. E. et al. Extensive transcriptional variation poses a challenge to thermal stress biomarker development for endangered corals. Mol. Ecol. 27, 1103–1119 (2018).
-
Christensen, K. A. et al. Assessing the effects of genotype-by-environment interaction on epigenetic, transcriptomic, and phenotypic response in a Pacific salmon. G3 11, jkab021 (2021).
-
Van Der Graaf, A. et al. Rate, spectrum, and evolutionary dynamics of spontaneous epimutations. Proc. Natl Acad. Sci. USA 112, 6676–6681 (2015).
-
Dixon, G., Liao, Y., Bay, L. K. & Matz, M. V. Role of gene body methylation in acclimatization and adaptation in a basal metazoan. Proc. Natl Acad. Sci. USA 115, 13342–13346 (2018).
-
Wu, Y. et al. Epigenome-wide association study of short-term temperature fluctuations based on within-sibship analyses in Australian females. Environ. Int. 171, 107655 (2023).
-
Oomen, R. A. & Hutchings, J. A. Genomic reaction norms inform predictions of plastic and adaptive responses to climate change. J. Anim. Ecol. 91, 1073–1087 (2022).
-
Ficetola, G. F., Miaud, C., Pompanon, F. & Taberlet, P. Species detection using environmental DNA from water samples. Biol. Lett. 4, 423–425 (2008).
-
Lacoursière-Roussel, A., Rosabal, M. & Bernatchez, L. Estimating fish abundance and biomass from eDNA concentrations: variability among capture methods and environmental conditions. Mol. Ecol. Resour. 16, 1401–1414 (2016).
-
Ruppert, K. M., Kline, R. J. & Rahman, M. S. Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: a systematic review in methods, monitoring, and applications of global eDNA. Glob. Ecol. Conserv. 17, e00547 (2019).
-
Clare, E. L. et al. eDNAir: proof of concept that animal DNA can be collected from air sampling. PeerJ 9, e11030 (2021).
-
Roger, F. et al. Airborne environmental DNA metabarcoding for the monitoring of terrestrial insects—a proof of concept from the field. Environ. DNA 4, 790–807 (2022).
-
Johnson, M. D., Fokar, M., Cox, R. D. & Barnes, M. A. Airborne environmental DNA metabarcoding detects more diversity, with less sampling effort, than a traditional plant community survey. BMC Ecol. Evol. 21, 218 (2021).
-
Machado, K. B. et al. DNA metabarcoding reveals the responses of prokaryotes and eukaryotes microbiota to warming: are the patterns similar between taxonomic and trophic groups? Ecol. Indic. 115, 106452 (2020). An experimental approach based on eDNA metabarcoding to evaluate the short-term effect of warming predicted by different future scenarios in the composition of the aquatic microbiota.
-
Ferguson, R. M. et al. The ecological impacts of multiple environmental stressors on coastal biofilm bacteria. Glob. Change Biol. 27, 3166–3178 (2021).
-
Moinet, G. Y. et al. Soil microbial sensitivity to temperature remains unchanged despite community compositional shifts along geothermal gradients. Glob. Change Biol. 27, 6217–6231 (2021). A natural long-term warming experiment based on eDNA metabarcoding to study switch in microbial community compositions depending on soil temperature.
-
Gallego, R., Jacobs-Palmer, E., Cribari, K. & Kelly, R. P. Environmental DNA metabarcoding reveals winners and losers of global change in coastal waters. Proc. Biol. Sci. 287, 20202424 (2020).
-
Djurhuus, A. et al. Environmental DNA reveals seasonal shifts and potential interactions in a marine community. Nat. Commun. 11, 254 (2020).
-
Abirami, B., Radhakrishnan, M., Kumaran, S. & Wilson, A. Impacts of global warming on marine microbial communities. Sci. Total Environ. 791, 147905 (2021).
-
Thurber, R. V. et al. Metagenomic analysis of stressed coral holobionts. Environ. Microbiol. 11, 2148–2163 (2009).
-
Busseni, G. et al. Large scale patterns of marine diatom richness: drivers and trends in a changing ocean. Glob. Ecol. Biogeogr. 29, 1915–1928 (2020).
-
Diner, R. E. et al. Pathogenic Vibrio species are associated with distinct environmental niches and planktonic taxa in Southern California (USA) aquatic microbiomes. mSystems 6, e0057121 (2021).
-
Wilcox, T. M. et al. Fine‐scale environmental DNA sampling reveals climate‐mediated interactions between native and invasive trout species. Ecosphere 9, e02500 (2018).
-
Isaak, D. J. et al. Do metapopulations and management matter for relict headwater bull trout populations in a warming climate? Ecol. Appl. 32, e2594 (2022).
-
Balint, M. et al. Environmental DNA time series in ecology. Trends Ecol. Evol. 33, 945–957 (2018).
-
Deiner, K. et al. Environmental DNA metabarcoding: transforming how we survey animal and plant communities. Mol. Ecol. 26, 5872–5895 (2017).
-
Willerslev, E. et al. Ancient biomolecules from deep ice cores reveal a forested southern Greenland. Science 317, 111–114 (2007).
-
Wang, Y. et al. Late quaternary dynamics of arctic biota from ancient environmental genomics. Nature 600, 86–92 (2021). A demonstration of the use of aDNA metagenomics to study how past climatic changes affected plant and animal communities.
-
Kjær, K. H. et al. A 2-million-year-old ecosystem in Greenland uncovered by environmental DNA. Nature 612, 283–291 (2022).
-
Zhang, H., Huo, S., Yeager, K. M. & Wu, F. Sedimentary DNA record of eukaryotic algal and cyanobacterial communities in a shallow Lake driven by human activities and climate change. Sci. Total Environ. 753, 141985 (2021).
-
Alsos, I. G. et al. Sedimentary ancient DNA from Lake Skartjørna, Svalbard: assessing the resilience of arctic flora to Holocene climate change. Holocene 26, 627–642 (2016).
-
Díaz, F. P. et al. Multiscale climate change impacts on plant diversity in the Atacama Desert. Glob. Change Biol. 25, 1733–1745 (2019).
-
Palkopoulou, E. et al. Holarctic genetic structure and range dynamics in the woolly mammoth. Proc. Biol. Sci. 280, 20131910 (2013).
-
Haile, J. et al. Ancient DNA reveals late survival of mammoth and horse in interior Alaska. Proc. Natl Acad. Sci. USA 106, 22352–22357 (2009).
-
Willerslev, E. et al. Fifty thousand years of Arctic vegetation and megafaunal diet. Nature 506, 47–51 (2014).
-
Thomsen, P. F. & Willerslev, E. Environmental DNA – an emerging tool in conservation for monitoring past and present biodiversity. Biol. Conserv. 183, 4–18 (2015).
-
Hechler, R. M., Yates, M. C., Chain, F. J. & Cristescu, M. E. Environmental transcriptomics under heat stress: can environmental RNA reveal changes in gene expression of aquatic organisms? Preprint at bioRxiv https://doi.org/10.1101/2022.10.06.510878 (2022).
-
Whitmore, L. et al. Inadvertent human genomic bycatch and intentional capture raise beneficial applications and ethical concerns with environmental DNA. Nat. Ecol. Evol. 7, 873–888 (2023).
-
Ficetola, G. F. & Taberlet, P. Towards exhaustive community ecology via DNA metabarcoding. Mol. Ecol. https://doi.org/10.1111/mec.16881 (2023).
-
Chen, Y. et al. The combination of genomic offset and niche modelling provides insights into climate change-driven vulnerability. Nat. Commun. 13, 4821 (2022).
-
Razgour, O. et al. Considering adaptive genetic variation in climate change vulnerability assessment reduces species range loss projections. Proc. Natl Acad. Sci. USA 116, 10418–10423 (2019).
-
Tournebize, R. et al. Ecological and genomic vulnerability to climate change across native populations of Robusta coffee (Coffea canephora). Glob. Change Biol. 28, 4124–4142 (2022).
-
Sherpa, S. et al. Genomic shifts, phenotypic clines, and fitness costs associated with cold tolerance in the Asian tiger mosquito. Mol. Biol. Evol. 39, 5 (2022).
-
Forester, B. R., Beever, E. A., Darst, C., Szymanski, J. & Funk, W. C. Linking evolutionary potential to extinction risk: applications and future directions. Front. Ecol. Environ. 20, 507–515 (2022).
-
Pan, H., Holbrook, J. D., Karnani, N. & Kwoh, C. K. Gene, environment and methylation (GEM): a tool suite to efficiently navigate large scale epigenome wide association studies and integrate genotype and interaction between genotype and environment. BMC Bioinformatics 17, 299 (2016).
-
Guo, X. et al. Linking genotype to phenotype in multi-omics data of small sample. BMC Genomics 22, 537 (2021).
-
Shi, W. J. et al. Unsupervised discovery of phenotype-specific multi-omics networks. Bioinformatics 35, 4336–4343 (2019).
-
Hanson, C., Cairns, J., Wang, L. & Sinha, S. Principled multi-omic analysis reveals gene regulatory mechanisms of phenotype variation. Genome Res. 28, 1207–1216 (2018).
-
Wellenreuther, M. & Bernatchez, L. Eco-evolutionary genomics of chromosomal inversions. Trends Ecol. Evol. 33, 427–440 (2018).
-
Layton, K. K. S. & Bradbury, I. R. Harnessing the power of multi-omics data for predicting climate change response. J. Anim. Ecol. 91, 1064–1072 (2022).
-
Euclide, P. et al. Is structural variation necessary to create islands of divergence in moderate gene flow species? A case study in sockeye salmon. Preprint at Authorea https://doi.org/10.22541/au.168371520.09492745/v1 (2023).
-
Wellenreuther, M., Mérot, C., Berdan, E. & Bernatchez, L. Going beyond SNPs: the role of structural genomic variants in adaptive evolution and species diversification. Mol. Ecol. 28, 1203–1209 (2019).
-
Hamann, E. et al. Review: plant eco-evolutionary responses to climate change: emerging directions. Plant Sci. 304, 110737 (2021).
-
Ferchiou, S., Caza, F., de Boissel, P. G. J., Villemur, R. & St-Pierre, Y. Applying the concept of liquid biopsy to monitor the microbial biodiversity of marine coastal ecosystems. ISME Commun. 2, 61 (2022).
-
Valdivieso, A., Anastasiadi, D., Ribas, L. & Piferrer, F. Development of epigenetic biomarkers for the identification of sex and thermal stress in fish using DNA methylation analysis and machine learning procedures. Mol. Ecol. Resour. 23, 453–470 (2023).
-
Horvath, S. & Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 19, 371–384 (2018).
-
Tumajer, J. et al. Forward modeling reveals multidecadal trends in cambial kinetics and phenology at treeline. Front. Plant Sci. 12, 613643 (2021).
-
Xuereb, A., Rougemont, Q., Tiffin, P., Xue, H. & Phifer-Rixey, M. Individual-based eco-evolutionary models for understanding adaptation in changing seas. Proc. Biol. Sci. 288, 20212006 (2021).
-
Carley, L. N., Morris, W. F., Walsh, R., Riebe, D. & Mitchell‐Olds, T. Are genetic variation and demographic performance linked? Evol. Appl. 15, 1888–1906 (2022).
-
Mathon, L. et al. The distribution of coastal fish eDNA sequences in the Anthropocene. Glob. Ecol. Biogeogr. 32, 1336–1352 (2023).
-
Lewin, H. A. et al. The earth BioGenome project 2020: starting the clock. Proc. Natl Acad. Sci. USA 119, e2115635118 (2022).
-
Jenkins, G. B. et al. Reproducibility in ecology and evolution: minimum standards for data and code. Ecol. Evol. 13, e9961 (2023).
-
Dauphin, B. et al. Re-thinking the environment in landscape genomics. Trends Ecol. Evol. 38, 261–274 (2023).
-
Waldvogel, A.-M. & Pfenninger, M. Temperature dependence of spontaneous mutation rates. Genome Res. 31, 1582–1589 (2021).
-
Leigh, D. M. et al. Opportunities and challenges of macrogenetic studies. Nat. Rev. Genet. 22, 791–807 (2021).
-
Schmidt, C., Hoban, S. & Jetz, W. Conservation macrogenetics: harnessing genetic data to meet conservation commitments. Trends Genet. https://doi.org/10.1016/j.tig.2023.08.002 (2023).
-
Hoban, S. et al. Global genetic diversity status and trends: towards a suite of essential biodiversity variables (EBVs) for genetic composition. Biol. Rev. 97, 1511–1538 (2022).
-
Hoban, S. et al. Genetic diversity goals and targets have improved, but remain insufficient for clear implementation of the post-2020 global biodiversity framework. Conserv. Genet. 24, 181–191 (2023).
-
Phillips, S. J., Anderson, R. P. & Schapire, R. E. Maximum entropy modeling of species geographic distributions. Ecol. Modell. 190, 231–259 (2006).
-
Hijmans, R. J., Cameron, S. E., Parra, J. L., Jones, P. G. & Jarvis, A. Very high resolution interpolated climate surfaces for global land areas. Int. J. Climatol. 25, 1965–1978 (2005).
-
McRae, B. H., Dickson, B. G., Keitt, T. H. & Shah, V. B. Using circuit theory to model connectivity in ecology, evolution, and conservation. Ecology 89, 2712–2724 (2008).
-
Excoffier, L. & Foll, M. Fastsimcoal: a continuous-time coalescent simulator of genomic diversity under arbitrarily complex evolutionary scenarios. Bioinformatics 27, 1332–1334 (2011).
-
Deatherage, D. E., Kepner, J. L., Bennett, A. F., Lenski, R. E. & Barrick, J. E. Specificity of genome evolution in experimental populations of Escherichia coli evolved at different temperatures. Proc. Natl Acad. Sci. USA 114, E1904–E1912 (2017).
-
Deatherage, D. E. & Barrick, J. E. Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseq. Methods Mol. Biol. 151, 165–188 (2014).
-
Lenski, R. E. Convergence and divergence in a long-term experiment with bacteria. Am. Nat. 190, S57–S68 (2017).
-
Jansen, M. et al. Thermal tolerance in the keystone species Daphnia magna — a candidate gene and an outlier analysis approach. Mol. Ecol. 26, 2291–2305 (2017).
Acknowledgements
This work was supported by an NSERC Discovery grant to L.B. C.J.V. is supported by an NSERC postdoctoral fellowship.
Author information
Authors and Affiliations
Contributions
The authors contributed equally to all aspects of the article.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Reviews Genetics thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
We wish to dedicate this review to the memory of Louis Bernatchez, who passed away shortly after its acceptance. We are all deeply grateful to Louis, not only for conceiving this piece of work, but most importantly for his outstanding contributions to the field, invaluable mentorship, and boundless generosity and compassion.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Glossary
- Assisted gene flow
-
Human-mediated movement of individuals within a species range to facilitate adaptation in recipient populations.
- Conservation macrogenetics
-
Studies of intraspecific genetic variation across many taxonomic groups and broad spatiotemporal scales to assess evolutionary patterns and inform conservation.
- Ecological niche model
-
(ENM). Sometimes referred to as environmental niche model or species distribution model. A class of methods that combines environmental data layers and species’ occurrence or abundance data to infer niche suitability and predict species’ distributions across current and future landscapes.
- Epigenome-wide association studies
-
(EWAS). Statistical method that links epigenomic variation to phenotypic or physiological outcomes.
- Evolve and resequence
-
(E&R). Integrates high-throughput sequencing into experimental evolution studies; most commonly performed on microbial and short-generation metazoans.
- Generalized dissimilarity modelling
-
(GDM). A statistical approach for prediction of nonlinear patterns of beta diversity across space; although initially developed for modelling turnover in community composition, it can also be applied to model genetic and trait differences.
- Genome scans
-
A genome-wide screening approach to identify candidate genomic variants (for example, SNPs) that are subject to selection.
- Genomic reaction norm
-
Graphical representations showing how a phenotype (generally gene expression, although other molecular phenotypes such as DNA methylation state can be used) of a single genotype changes in response to different environments.
- Individual-based modelling
-
(IBM). Modelling approach in which the actions of individual organisms are simulated, including interactions with other individuals and with the environment.
- Integral projection models
-
Models that predict the effects of individuals’ traits (for example, age, reproductive success, growth rate, body size) on population size and demography.
- Landscape genomics
-
The study of the spatial distribution of genome-wide variation, including both neutral and adaptive variation, across heterogeneous landscapes and the processes that shape observed distributions.
- Landscape resistance
-
A measure of the difficulty for organisms to move through the landscape; areas with high resistance can impede dispersal and gene flow.
- Liquid biopsy
-
Noninvasive test to detect noncellular DNA in bodily fluids, often used for cancer and disease detection.
- Local adaptation
-
Mechanism by which organisms in a population evolve phenotypes that have higher relative fitness in their home conditions compared with organisms in other populations in spatially heterogeneous environments.
- Nonsense substitutions
-
Point mutations in a sequence of DNA that result in a premature stop codon.
- Non-synonymous substitutions
-
Nucleotide mutation that alter the amino acid sequence of a protein.
- Otoliths
-
Calcium carbonate structures in the inner ear of vertebrates.
- Quantitative trait loci
-
(QTLs). Genetic variants associated with changes in expression (eQTL) or DNA methylation (methylQTL) levels.
- Rescue effects
-
An increase in population size or fitness, thereby reducing the risk of extinction and facilitating long-term population persistence. Three eco-evolutionary processes promote rescue effects: demographic rescue, that is, an increase in the number of individuals in a population; genetic rescue, that is, an increase in population fitness via the genetic contribution of immigrants, by either decreasing inbreeding depression or increasing genetic variation to promote adaptation; evolutionary rescue, that is, an increase in population growth rates caused by adaptation to new or changing conditions, usually from standing genetic variation but some definitions include migration.
- Standing genetic variation
-
Pre-existing genetic variation in a population on which selection can act readily.
- Structural variation
-
A region of the genome that shows inter-individual variability due to a chromosomal alteration, including variation in chromosomal location (for example, translocation), rearrangement of chromosome orientation (for example, inversion) or copy number variation (for example, duplications, insertions, deletions).
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article
Cite this article
Bernatchez, L., Ferchaud, AL., Berger, C.S. et al. Genomics for monitoring and understanding species responses to global climate change.
Nat Rev Genet (2023). https://doi.org/10.1038/s41576-023-00657-y
-
Accepted: 29 August 2023
-
Published: 20 October 2023
-
DOI: https://doi.org/10.1038/s41576-023-00657-y