Abstract
Synthetic carbon allotropes such as graphene1, carbon nanotubes2 and fullerenes3 have revolutionized materials science and led to new technologies. Many hypothetical carbon allotropes have been discussed4, but few have been studied experimentally. Recently, unconventional synthetic strategies such as dynamic covalent chemistry5 and on-surface synthesis6 have been used to create new forms of carbon, including γ-graphyne7, fullerene polymers8, biphenylene networks9 and cyclocarbons10,11. Cyclo[N]carbons are molecular rings consisting of N carbon atoms12,13; the three that have been reported to date (N = 10, 14 and 18)10,11 are doubly aromatic, which prompts the question: is it possible to prepare doubly anti-aromatic versions? Here we report the synthesis and characterization of an anti-aromatic carbon allotrope, cyclo[16]carbon, by using tip-induced on-surface chemistry6. In addition to structural information from atomic force microscopy, we probed its electronic structure by recording orbital density maps14 with scanning tunnelling microscopy. The observation of bond-length alternation in cyclo[16]carbon confirms its double anti-aromaticity, in concordance with theory. The simple structure of C16 renders it an interesting model system for studying the limits of aromaticity, and its high reactivity makes it a promising precursor to novel carbon allotropes15.
Main
Many cyclo[N]carbons (N = 6–40) have been detected in the gas phase12,13,16, and two examples (C6 and C8) have been trapped in solid argon and characterized by infrared spectroscopy17,18. Cyclo[10]carbon, cyclo[14]carbon and cyclo[18]carbon have been characterized by scanning probe microscopy of individual molecules on NaCl surfaces at low temperature10,11,19. Atomic force microscopy (AFM) images revealed cumulenic structures for C10 and C14 with bond-angle alternation (BAA)11 and a polyynic structure for C18 (refs. 10,19). Cyclo[N]carbons with N = 4n + 2 (where n is an integer), such as C10, C14 and C18, are expected to be doubly aromatic and to have special stability, due to their closed-shell electronic configurations, relating to the presence of in-plane and out-of-plane aromatic Hückel circuits of 4n + 2 π electrons20,21,22,23,24,25. By contrast, cyclo[4n]carbons have been predicted to be less stable and doubly anti-aromatic22,23,24,25,26. Here we report the first structural characterization of a cyclo[4n]carbon to our knowledge. C16 was prepared on a NaCl surface by tip-induced chemistry from a C16(CO)4Br2 precursor. AFM and scanning tunnelling microscopy (STM) provide insight into the geometry and electronic structure, respectively, of neutral C16 and anionic C16–. We find that neutral C16 exhibits significant bond-length alternation (BLA), which confirms its double anti-aromaticity. Our experimental results are complemented by state-of-the-art quantum mechanical calculations, as well as by methods suitable for execution on a quantum computer.
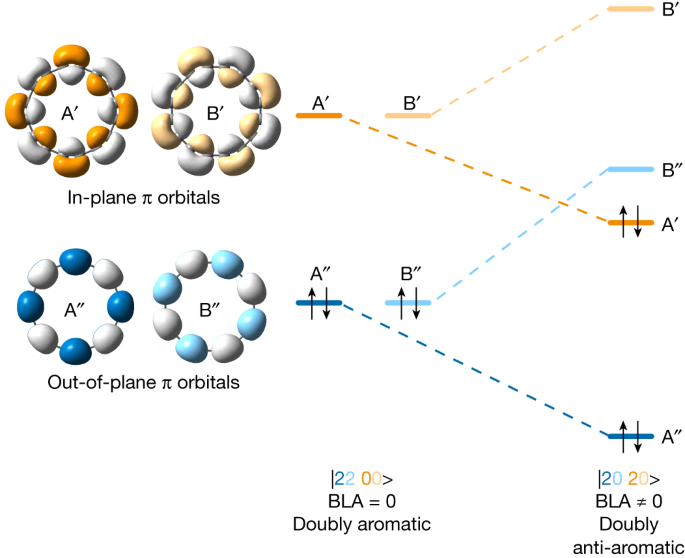
Cyclocarbons have two orthogonal π systems, one with orbital lobes in the ring plane and the other out of plane, with nodes in the ring plane. In an infinitely large cyclocarbon, these two π systems are degenerate; but in a finite ring, in-plane frontier orbitals are slightly higher in energy than their out-of-plane counterparts13. This pattern of orbitals can lead to several possible electronic states. In the D16h geometry of C16 with no BLA, the ground state may be a doubly aromatic |2200> state (Fig. 1, left), with 18 (4n + 2) and 14 (4n − 2) electrons in out-of-plane and in-plane π systems, respectively. In this state, there are two degenerate pairs of frontier orbitals (out-of-plane A′′ and B′′ are occupied, and in-plane A′ and B′ are unoccupied). If we introduce BLA (D8h symmetry), these orbital pairs cease to be degenerate, with one member of each pair (A in Fig. 1, right) becoming stabilized relative to the other (B). This symmetry breaking leads to a doubly anti-aromatic |2020> configuration with 16 electrons in both in-plane and out-of-plane π systems. A third possible state would be |1111> , with D16h symmetry, but such open-shell configurations are known to be unstable relative to closed-shell alternatives27.

In-plane orbitals are labelled A′ and B′ and out-of-plane orbitals A′′ and B′′. Orbitals A′, B′ (and A′′, B′′) are related by rotation and have equal energy when all bonds are of equal length. Introducing BLA lifts this degeneracy, resulting in orbital reordering and a doubly anti-aromatic ground state.
The unique structure, small size and high symmetry of cyclocarbons has made them a target of many theoretical studies, sometimes producing conflicting results13. Here, we investigate C16 using both state-of-the-art computational methods and a variational quantum eigensolver28 paired with the quantum unitary coupled-cluster singles and doubles (q-UCCSD)29 ansatz. These calculations confirm that the doubly anti-aromatic configuration is the ground state of C16, with strong BLA.
Precursor synthesis
Cyclo[16]carbon was synthesized as shown in Fig. 2. Glaser–Hay coupling of a mixture of alkynes 1 and 2 gave macrocycle 3 in 20% yield, and the structure of this product was confirmed by single-crystal X-ray diffraction (Supplementary Fig. 3). Compounds 3 and 4 are anti-aromatic (as confirmed by the 1H nuclear magnetic resonance spectrum of compound 3; Supplementary Fig. 2). Deprotection of 3 to give 4 proved difficult because of the high reactivity of compound 4, but after testing many reaction conditions, we found that 3 can be converted to 4 in 94% yield using trifluoroacetic acid containing water (2.5% by volume).

TMEDA is tetramethylethylenediamine.
On-surface synthesis and characterization
Precursor 4 was sublimed by fast heating from a Si wafer10 onto a Cu(111) single-crystal surface partially covered with NaCl at a sample temperature of about T = 10 K. On-surface synthesis (Fig. 3a) and characterization by STM and AFM with CO-tip functionalization30,31 were performed at T = 5 K. We found intact molecules of 4 on bilayer NaCl, denoted NaCl(2 ML)/Cu(111), as shown in Fig. 3b. The Br atoms appear as bright (repulsive) dots in the AFM image19, whereas the CO masking groups are dark features10. The triple bonds show up as bright features due to bond-order related contrast obtained with CO-tip functionalization10,31,32 (for further data on 4, see Supplementary Fig. 6).

a, Reaction scheme. b–e, Constant-height, CO-tip AFM images of precursor 4 (b), intermediates 5 (c) and 6 (d), and C16 (e). f–i, AFM image of C16 in neutral (f) and anionic (g) charge state, and calculated lowest-energy adsorption sites of C160 (h) and C16– (i) on NaCl (colour code: Na pink, Cl green). j–m, C160 adsorbed in a bay of a third-layer NaCl island, imaged with AFM at different decreasing tip-height offsets: +0.20 Å (j), +0 Å (k), −0.45 Å (l) and −0.55 Å (m). All molecules are adsorbed on NaCl(2 ML)/Cu(111). The tip-height offsets provided in the images refer to the STM setpoint of I = 0.2 pA and V = 0.2 V on bare NaCl(2 ML)/Cu(111). Scale bars, 0.5 nm.
Voltage pulses applied for a few seconds at constant tip height were used to unmask the acetylenes in individual molecules of precursor 4. We successively increased the voltage and decreased the tip height for the pulse until it resulted in dissociation reactions. For tunnelling currents on the order of few pA, the minimum voltage required for debromination of 4 to give 5 (Fig. 3c; see Supplementary Fig. 7 for further data) was 1.3 V, coinciding with the bias for resonant tunnelling: that is, electron attachment to 4 (Supplementary Fig. 6). For CO unmasking, larger bias voltages were required, typically about 3 V. We speculate that the dissociation reactions are triggered in transiently charged species by inelastic electron tunnelling processes31. Intermediate 6 was observed after dissociating the first pair of CO masking groups (Fig. 3d; see Supplementary Fig. 8 for further data). Removal of a second pair of CO molecules gave the final product, C16 (Fig. 3e and Supplementary Figs. 9 and 10). Previously, gas-phase C16 has been formed from a molecular precursor33,34 and studied in its anionic34,35 and cationic16,36 forms, but to our knowledge, this is the first time C16 has been generated in a condensed phase or structurally characterized. The yield for the on-surface synthesis of C16 was about 30%; in unsuccessful attempts, the ring opened to form linear polyynic chains (Supplementary Fig. 11) or the molecule was picked up by the tip.
We observed C16 in two different forms on the NaCl surface (Fig. 3f,g) that we assign to neutral C160 and negatively charged C16–, respectively (see also Fig. 4, Supplementary Figs. 12 and 13 and Supplementary Tables 1 and 2). Whereas C160 appears circular, C16– adopts a distorted oval geometry. We observed a variety of adsorption sites for C160 on the NaCl surface (Supplementary Fig. 14), indicating a weak interaction with the substrate. In contrast, C16– showed a systematic preference for adsorption above a bridge site (Supplementary Figs. 15 and 16). To investigate the interaction of C160 and C16– with the NaCl surface, we performed density functional theory (DFT) calculations with periodic boundary conditions, both on a pristine surface and at NaCl island step edges. The calculated lowest-energy adsorption sites of C160 and C16– on pristine NaCl are shown in Fig. 3h,i, respectively. For the neutral charge state, we calculated an adsorption energy of 0.65 eV, similar to the value of 0.67 eV previously calculated for C18 on NaCl (ref. 37) that was predicted to diffuse freely across the surface even at low temperatures. The calculated relaxed adsorption geometry of C16– on pristine NaCl is oval shaped, with the molecule centred on a bridge site (Fig. 3i), in agreement with its experimentally observed site and shape (Fig. 3g, Supplementary Fig. 15 and Supplementary Table 1). This adsorption geometry can be attributed to electrostatic interactions of the C16– anion with the Na cations and Cl anions, resulting in a substantially stronger adsorption energy (1.44 eV) than that of the neutral molecule.

a,b, Constant-current STM images of C16 in neutral (a) and negative charge state (b), respectively (V = 50 mV, I = 0.2 pA). c, Difference of panels b and a. d,e, Constant-height AFM images of C160 (d) and C16– (e). f, Constant-current STM (I = 0.4 pA and V = +1.2 V) mapping the ionic resonance of C16– to C162–. g–i, Same data as d–f after applying a Laplace filter. The molecule was adsorbed on NaCl(2 ML)/Cu(111) near a third-layer island. Scale bars, 0.5 nm. k,l, Optimized geometries (ωB97XD/def2-TZVP) of C160 (k) and C16– (l), with bond lengths and bond angles indicated. m, Simulated isosurface at 0.2 atomic units (1.4 e/Å–3) of the LUMO of C16–.
The C16 molecules frequently moved on the surface during imaging with AFM and STM, indicating a small diffusion barrier and making them challenging to characterize. We never observed neutral C16 stably isolated on the NaCl surface, but always near a third-layer NaCl step edge, to provide a more stable adsorption site and facilitate detailed characterization. Figure 3j–m shows C16 adsorbed in a bay of a third-layer island imaged with AFM at different tip heights. Kelvin probe force spectroscopy confirmed that the molecule in Fig. 3f,j–m is charge neutral (Supplementary Tables 2 and 3 and Supplementary Fig. 17). The bright contrast obtained by CO-tip AFM above the triple bonds for larger tip heights (Fig. 3j,k) evolves to the shape of an octagon with corners at the positions of triple bonds at decreased tip heights (Fig. 3l,m). The results indicate BLA10: that is, a polyynic structure of neutral C16. Our computations (Supplementary Table 1 and Supplementary Figs. 54 and 55) predict a larger adsorption energy (1.13 and 2.61 eV for C160 and C16–, respectively) at defect sites compared to the pristine surface, accompanied by an increase in BAA (up to 35° for C160 and up to 50° for C16–, compared to 20–30° on a pristine surface). BLA is maintained in all cases, with no fundamental changes in the electronic structure.
Charge-state switching
The charge state of C16 can be controllably switched using the applied bias, as shown in Fig. 4. At about V = 0.5 V, the molecule switched from neutral C160 to the anion C16–, (and at V = –0.3 V in the reverse direction, C16– to C160; Supplementary Figs. 12 and 18). The STM images in Fig. 4a,b show C160 and C16–, respectively. The negative charge state leads to a characteristic dark halo (Fig. 4b) and interface state scattering as observed in the difference image Fig. 4c (ref. 38); see also Supplementary Fig. 12 for images with enhanced contrast. The assignments of these charge states are corroborated by Kelvin probe force spectroscopy (Supplementary Fig. 18). AFM data for C160 and C16– are shown in Fig. 4d,e with corresponding Laplace-filtered data in Fig. 4g,h, respectively. In this case, the structural distortion of C160 and C16– is similar, which we assign to the influence of the third-layer NaCl island (Supplementary Table 1).
The more stable adsorption at the third-layer island allowed us to image the molecule at increased bias voltages without inducing movement of the molecule. At about 1.2 V, we observe the onset of an electronic resonance by scanning tunnelling spectroscopy (Supplementary Fig. 18). The STM image at 1.2 V shown in Fig. 4f (Laplace-filtered data in Fig. 4i), reveals the orbital density corresponding to that resonance14. As the molecule is already in the anionic charge state at V > 0.5 V, we assign this resonance to the transition from anionic C16– to the dianionic charge state C162–, giving us insight into the electronic structure of C16.
Multireference methods and DFT (see Supplementary Tables 4 and 5 for details) both predict C160 to have a |2020> ground state with a polyynic geometry and BLA, but no BAA, in the gas phase (D8h symmetry, Fig. 4k). The electronic structure of C160 (Fig. 1, right, and Supplementary Fig. 19) features a nearly degenerate pair of highest occupied molecular orbitals A′′ (HOMO–1) and A′ (HOMO), as well a nearly degenerate pair of lowest unoccupied molecular orbitals B′′ (LUMO) and B′ (LUMO + 1). A′ and B′ (as well as A′′ and B′′) are related by rotation; in a magnetic field, they couple to induce a strong ring current (–25 nA T–1, cf. 12 nA T–1 in benzene), reinforcing the applied field inside the ring. This current is a signature of anti-aromaticity39, and it can be visualized by nucleus-independent chemical shift calculations (see comparison of the plots for the |2200> and |2020> states of C16 in Supplementary Fig. 28). In contrast to the neutral |2020> state of C16, the anion shows both BLA and BAA (Fig. 4l), due to single occupation of the B′′ orbital, resulting in C8h symmetry.
The DFT-predicted LUMO of C16– (Fig. 4m and Supplementary Fig. 20) can be compared to the electronic resonance imaged by STM (Fig. 4f,i), which corresponds to the squared orbital wavefunction14,38, and to the addition of a second electron to the singly occupied out-of-plane orbital (B′′) in C16–. Both theory and experiment show high-density lobes above the long bonds of C16–, which are located between the bright features of the corresponding AFM images. The symmetry lowering from D8h to C8h in C16–, which is the effect of BAA, is reflected in the shape of the orbital lobes and can be observed in both experiment (Fig. 4f,i) and theory (Fig. 4m). AFM data showing BLA, and STM data showing the orbital density for the C16– to C162– transition, corresponding to the addition of an electron to the B′′ orbital of C16–, are all in excellent agreement with the calculations, strongly indicating the doubly anti-aromatic character of C160, which causes pronounced BLA and a D8h geometry. The two other possible electronic configurations of C16, doubly aromatic |2200> and open-shell |1111>, were calculated by DFT to have nearly identical D16h minima with no BLA and substantially higher energies (2.47 and 1.78 eV, respectively) than the doubly anti-aromatic |2020> ground state. Relative ground-state energies of the D8h and D16h minima were also determined using q-UCCSD by simulating quantum circuits with Qiskit40. q-UCCSD predicts that the D8h minimum is more stable than the D16h minimum by 3.38 eV, which is very similar to the result obtained using conventional coupled-cluster singles and doubles (3.31 eV; see Supplementary Information for further discussion).
Our experimental results, most importantly the observed BLA for neutral C16, confirm the occupation of both π systems (in-plane and out-of-plane) with 16 electrons, making the molecule doubly anti-aromatic. Ring current calculations on neutral C16 also indicate significant anti-aromaticity in this electronic configuration. The investigation of both C160 and C16– provides confidence in the assignment of charge states and insights into the electronic structure of the molecule. The synthesis, stabilization and characterization of C16 opens the way to create other elusive carbon-rich anti-aromatic molecules by atom manipulation.
Data availability
The data that support the findings of this study are available in the paper and its Supplementary Information, or are available from the Zenodo public repository (https://zenodo.org/record/8226451 and https://doi.org/10.5281/zenodo.8226451). Crystallographic data for compound 3 are available free of charge from the Cambridge Crystallographic Data Centre (CCDC 2240722), https://www.ccdc.cam.ac.uk/data_request/cif.
References
-
Novoselov, K. S. et al. Electric field effect in atomically thin carbon films. Science 306, 666–669 (2004).
Google Scholar
-
Iijima, S. & Ichihashi, T. Single-shell carbon nanotubes of 1-nm diameter. Nature 363, 603–605 (1993).
Google Scholar
-
Kroto, H. W., Heath, J. R., O’Brien, S. C., Curl, R. F. & Smalley, R. E. C60: buckminsterfullerene. Nature 318, 162–163 (1985).
Google Scholar
-
Zhang, R.-S. & Jiang, J.-W. The art of designing carbon allotropes. Front. Phys. 14, 13401 (2019).
Google Scholar
-
Diercks, C. S. & Yaghi, O. M. The atom, the molecule, and the covalent organic framework. Science 355, eaal1585 (2017).
Google Scholar
-
Clair, S. & de Oteyza, D. G. Controlling a chemical coupling reaction on a surface: tools and strategies for on-surface synthesis. Chem. Rev. 119, 4717–4776 (2019).
Google Scholar
-
Hu, Y. et al. Synthesis of γ-graphyne using dynamic covalent chemistry. Nat. Synth. 1, 449–454 (2022).
Google Scholar
-
Meirzadeh, E. et al. A few-layer covalent network of fullerenes. Nature 613, 71–76 (2023).
Google Scholar
-
Fan, Q. et al. Biphenylene network: a nonbenzenoid carbon allotrope. Science 372, 852–856 (2021).
Google Scholar
-
Kaiser, K. et al. An sp-hybridized molecular carbon allotrope, cyclo[18] carbon. Science 365, 1299–1301 (2019).
Google Scholar
-
Sun, L. et al. Aromatic annular carbon allotropes: cumulenic cyclo[10]carbon and Peierls-transition-intermediate cyclo[14]carbon. Preprint at https://www.researchsquare.com/article/rs-2616838/v2 (2023).
-
Tobe, Y. & Wakabayashi, T. in Polyynes: Synthesis, Properties, and Applications (ed. Cataldo, F.) Ch. 6 (CRC/Taylor & Francis, 2006).
-
Anderson, H. L., Patrick, C. W., Scriven, L. M. & Woltering, S. L. A short history of cyclocarbons. Bull. Chem. Soc. Jpn 94, 798–811 (2021).
Google Scholar
-
Repp, J., Meyer, G., Stojkovic, S. M., Gourdon, A. & Joachim, C. Molecules on insulating films: scanning-tunneling microscopy imaging of individual molecular orbitals. Phys. Rev. Lett. 94, 26803 (2005).
Google Scholar
-
Diederich, F. Carbon scaffolding: building acetylenic all-carbon and carbon-rich compounds. Nature 369, 199–207 (1994).
Google Scholar
-
Marlton, S. J. P. et al. Probing colossal carbon rings. J. Phys. Chem. A 127, 1168–1178 (2023).
Google Scholar
-
Wang, S. L., Rittby, C. M. L. & Graham, W. R. M. Detection of cyclic carbon clusters. I. Isotopic study of the ν4(e’) mode of cyclic C6 in solid Ar. J. Chem. Phys. 107, 6032–6037 (1997).
Google Scholar
-
Wang, S. L., Rittby, C. M. L. & Graham, W. R. M. Detection of cyclic carbon clusters. II. Isotopic study of the ν12(eu) mode of cyclic C8 in solid Ar. J. Chem. Phys. 107, 7025–7033 (1997).
Google Scholar
-
Scriven, L. M. et al. Synthesis of cyclo[18] carbon via debromination of C18Br6. J. Am. Chem. Soc. 142, 12921–12924 (2020).
Google Scholar
-
Diederich, F. et al. All-carbon molecules: evidence for generation of cyclo[18] carbon from a stable organic precursor. Science 245, 1088–1090 (1989).
Google Scholar
-
Schleyer, P. V. R., Jiao, H., Glukhovtsev, M. N., Chandrasekhar, J. & Kraka, E. Double aromaticity in the 3,5-dehydrophenyl cation and in cyclo[6]carbon. J. Am. Chem. Soc. 116, 10129–10134 (1994).
Google Scholar
-
Fowler, P. W., Mizoguchi, N., Bean, D. E. & Havenith, R. W. A. Double aromaticity and ring currents in all-carbon rings. Chem. Eur. J. 15, 6964–6972 (2009).
Google Scholar
-
Charistos, N. D. & Muñoz-Castro, A. Induced magnetic field in sp-hybridized carbon rings: analysis of double aromaticity and antiaromaticity in cyclo[2N]carbon allotropes. Phys. Chem. Chem. Phys. 22, 9240–9249 (2020).
Google Scholar
-
Baryshnikov, G. V. et al. Aromaticity of even-number cyclo[n]carbons. J. Phys. Chem. A 124, 10849–10855 (2020).
Google Scholar
-
Hutter, J., Lüthi, H. P. & Diederich, F. Structures and vibrational frequencies of the carbon molecules C2-C18 calculated by density functional theory. J. Am. Chem. Soc. 116, 750–756 (1994).
Google Scholar
-
Ohno, K. Quantum chemical exploration of conversion pathways and isomeric structures of C16 molecules. Chem. Phys. Lett. 711, 60–65 (2018).
Google Scholar
-
Wenthold, P. G., Hrovat, D. A., Borden, W. T. & Lineberger, W. C. Transition-state spectroscopy of cyclooctatetraene. Science 272, 1456–1459 (1996).
Google Scholar
-
Peruzzo, A. et al. A variational eigenvalue solver on a photonic quantum processor. Nat. Commun. 5, 4213 (2014).
Google Scholar
-
Barkoutsos, P. K. et al. Quantum algorithms for electronic structure calculations: particle-hole Hamiltonian and optimized wave-function expansions. Phys. Rev. A 98, 022322 (2018).
Google Scholar
-
Gross, L., Mohn, F., Moll, N., Liljeroth, P. & Meyer, G. The chemical structure of a molecule resolved by atomic force microscopy. Science 325, 1110–1114 (2009).
Google Scholar
-
Pavliček, N. et al. Polyyne formation via skeletal rearrangement induced by atomic manipulation. Nat. Chem. 10, 853–858 (2018).
Google Scholar
-
Gross, L. et al. Bond-order discrimination by atomic force microscopy. Science 337, 1326–1329 (2012).
Google Scholar
-
Tobe, Y., Matsumoto, H., Naemura, K., Achiba, Y. & Wakabayashi, T. Generation of cyclocarbons with 4n carbon atoms (C12, C16, and C20) by [2 + 2] cycloreversion of propellane-annelated dehydroannulenes. Angew. Chem. Int. Edn 35, 1800–1802 (1996).
Google Scholar
-
Wakabayashi, T. et al. Photoelectron spectroscopy of Cn− produced from laser ablated dehydroannulene derivatives having carbon ring size of n = 12, 16, 18, 20, and 24. J. Chem. Phys. 107, 4783–4787 (1997).
Google Scholar
-
Ohara, M., Kasuya, D., Shiromaru, H. & Achiba, Y. Resonance-enhanced multiphoton electron detachment (REMPED) study of carbon anions up to C21–. J. Phys. Chem. A 104, 8622–8626 (2000).
Google Scholar
-
von Helden, G., Hsu, M.-T., Kemper, P. R. & Bowers, M. T. Structures of carbon cluster ions from 3 to 60 atoms: linears to rings to fullerenes. J. Chem. Phys. 95, 3835–3837 (1991).
Google Scholar
-
Baryshnikov, G. V., Valiev, R. R., Kuklin, A. V., Sundholm, D. & Ågren, H. Cyclo[18]carbon: insight into electronic structure, aromaticity, and surface coupling. J. Phys. Chem. Lett. 10, 6701–6705 (2019).
Google Scholar
-
Repp, J., Meyer, G., Olsson, F. E. & Persson, M. Controlling the charge state of individual gold adatoms. Science 305, 493–495 (2004).
Google Scholar
-
Gershoni-Poranne, R. & Stanger, A. Magnetic criteria of aromaticity. Chem. Soc. Rev. 44, 6597–6615 (2015).
Google Scholar
-
Anis, M. S. et al. Qiskit: an open-source framework for quantum computing. Zenodo https://doi.org/10.5281/zenodo.2573505 (2021).
Acknowledgements
We thank the following organizations for support: European Research Council grant no. 885606, ARO-MAT (H.L.A., Y.G.); European Community Horizon 2020 grant project 101019310 CycloCarbonCatenane (Y.G., H.L.A.); European Community grant ElDelPath (I.R., H.L.A.); Leverhulme Trust (Project Grant RPG-2017-032) (H.L.A., L.M.S.); European Research Council Synergy grant MolDAM (grant no. 951519); and European Union project SPRING (grant no. 863098). Computational resources were provided by Cirrus UK National Tier-2 HPC Service at EPCC (http://www.cirrus.ac.uk), funded by the University of Edinburgh and EPSRC (EP/P020267/1); and the Ministry of Education, Youth and Sports of the Czech Republic through the e-INFRA CZ (ID:90140). IBM, the IBM logo, and ibm.com are trademarks of the International Business Machines Corp., registered in many jurisdictions worldwide. Other product and service names might be trademarks of IBM or other companies. The current list of IBM trademarks is available at https://www.ibm.com/legal/copytrade.
Author information
Authors and Affiliations
Contributions
H.L.A. and L.G. conceived and initiated the project. Y.G., I.E., P.K. and L.M.S. synthesized the precursors. F.A. and L.G. carried out the atom manipulation and scanning probe microscopy. I.R., L.R., M.R. and I.T. performed theoretical analysis and computational simulations. K.E.C. determined the crystal structure of compound 3. Y.G., F.A., I.R., H.L.A. and L.G. wrote the paper. All authors discussed the results and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature thanks the anonymous reviewers for their contribution to the peer review of this work. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary experimental and computational methods, figures and data.
Peer Review File
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
Reprints and Permissions
About this article
Cite this article
Gao, Y., Albrecht, F., Rončević, I. et al. On-surface synthesis of a doubly anti-aromatic carbon allotrope.
Nature (2023). https://doi.org/10.1038/s41586-023-06566-8
-
Received: 14 April 2023
-
Accepted: 23 August 2023
-
Published: 25 October 2023
-
DOI: https://doi.org/10.1038/s41586-023-06566-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.