Abstract
New molecular technologies have helped unveil previously unexplored facets of the genome beyond the canonical proteome, including microproteins and short ORFs, products of alternative splicing, regulatory non-coding RNAs, as well as transposable elements, cis-regulatory DNA, and other highly repetitive regions of DNA. In this Review, we highlight what is known about this ‘hidden genome’ within the fungal kingdom. Using well-established model systems as a contextual framework, we describe key elements of this hidden genome in diverse fungal species, and explore how these factors perform critical functions in regulating fungal metabolism, stress tolerance, and pathogenesis. Finally, we discuss new technologies that may be adapted to further characterize the hidden genome in fungi.
Introduction
The canonical human proteome represents a universally recognized and comprehensive set of proteins encoded by the human genome, and its establishment has been critical to understanding fundamental cellular processes1. However, traditional annotations of this canonical proteome often overlook the polycistronic nature of genes and the factors encoded by non-canonical open reading frames (ORFs) that exist within and outside of the primarily recognized protein-coding genome2,3. Indeed, the combination of many non-ORF genomic features, including microproteins encoded within and adjacent to larger canonical protein sequences, non-coding RNA and DNA, and products of alternative splicing, all contribute to the emerging concept of the ‘hidden genome’ (Fig. 1). For example, in human cell lines, many unique proteins with differing functions can be encoded within a single gene sequence2,4. This growing appreciation of the nested and entangled nature of ORFs in the translatome has demonstrated that our traditional understanding of the functional potential within cells has been historically underestimated5. Our understanding of cellular function is further complicated by the classification of features such as long non-coding RNAs, which can function as RNAs, though, other times, actually do encode proteins critical to cell function6,7. Non-coding DNA has become of particular interest since genome-wide association studies suggest that over 90% of human disease-associated DNA variants are found in the non-coding genome8. Certainly, defining the canonical human proteome appears to have been just the beginning of characterizing the many intricate genetic products that contribute to cellular fitness in the human cell.
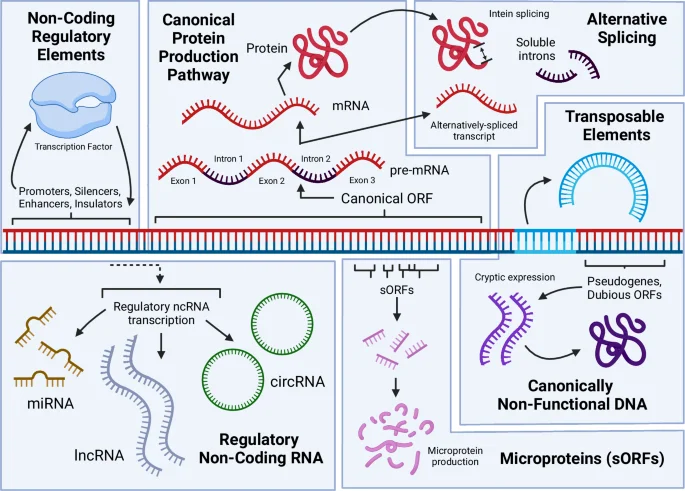
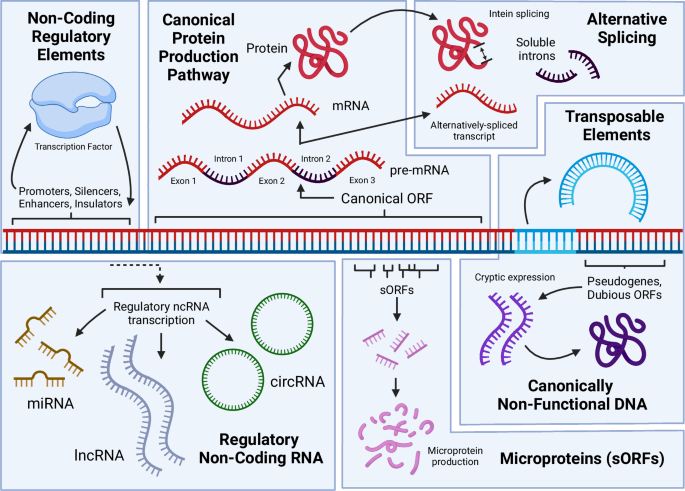
Sources of function from DNA include: production of mRNA and the resulting canonical proteins that are the focus of most genomic research; alternatively spliced RNA and protein products, including introns that are capable of becoming fixed in a cell and regulating gene expression; ubiquitous transcription of diverse non-coding RNAs that play important roles via their interaction with DNA, RNA, and proteins; the expression of pseudogenized DNA and dubious ORFs into functional proteins; the existence of non-coding regulatory elements (NCREs) that regulate gene expression; transposable elements that may move within the genome or between cells within or outside of the originating species, along with other repetitive DNA that may facilitate structural rearrangements in the genome. Figure 1, created with BioRender.com, released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license.
This phenomenon of the hidden genome extends across the tree of life and includes fungal species, where non-ORF genes that contribute to fitness have traditionally been neglected and are only recently beginning to be identified and characterized9,10. Indeed, the genomes of many fungal species important to human health and disease, agriculture, and biotechnology, remain incompletely characterized11. There is substantial emerging evidence for a critical role of the hidden genome, including microproteins and alternatively spliced proteins, microRNAs (miRNAs), long non-coding RNAs (lncRNAs), and circular RNAs (circRNAs), as well as other regions of non-coding and repetitive DNA, in numerous key facets of fungal biology (Table 1). Therefore, the ability to fully characterize these cryptic components of the fungal genome will be a critical step towards a comprehensive understanding of the genomics and biology of the fungal kingdom. The fact that genomic tools generally take longer to adapt and utilize in fungi has meant that many components of the hidden genome have gone relatively underexplored compared to other model species, though advances in other model organisms do offer a glimpse into how new tools may be employed in fungi. As the vast diversity of the fungal kingdom plays significant roles in all aspects of human life, it is imperative to fully dissect the functional potential of these fungal genomes.
Here, we describe the emergent findings that have been made into the hidden genome of diverse fungal species. We describe key components of the genome outside the canonical proteome, using both fungal and other species as relevant context, describe the important role of these factors in mediating important facets of fungal biology, and discuss new technologies that may be adapted to study the hidden genome in fungal species.
Non-canonical ORFs
Microproteins
Microproteins are proteins made up of a polypeptide chain shorter than 100 amino acids in length, encoded by short open reading frames (sORFs)12. These proteins were originally omitted from most functional analysis research due to the expectation that they would only rarely impact fitness, and for practical reasons due to their massive abundance in the genome12. However, the use of newer, more sensitive technologies has allowed for the confident detection of thousands of microproteins translated from sORFs in human cells, many of which have been characterized as playing important roles, including their involvement in stress response pathways13. Foundational studies in characterizing microprotein function in human cell lines have revealed the potentially profound impact of the microproteome on the development of disease14. In bacteria, many microproteins have been implicated in drug resistance, as well as in toxin-antitoxin systems and oxidative stress15.
Microproteins encoded by sORFs have also been recognized as having putative regulatory functions in fungi for decades, though, by their nature, it is difficult to distinguish between sORFs that are transcribed and translated and those randomly occurring incidental small ORFs that are not16. Regardless, bioinformatic approaches for annotating genomic sORFs have improved substantially over the past few years, and have been used to predict thousands of sORFs in 31 different fungal genomes17. One study in Saccharomyces cerevisiae leveraged ribosome profiling datasets to determine that translation of non-canonical ORFs may occur within the DNA sequences of at least 15% of canonical ORF-encoding genes18. Hypothetical annotations such as these can serve as the foundation for experimental approaches while attempting to successfully identify microproteins. One combinatorial method using ribosome profiling and proteomics, for example, was used in the fission yeast Schizosaccharomyces pombe to verify the existence of peptides corresponding to hypothesized sORFs19. However, in this case, only 9/373 of the presumed sORFs had a detectable peptide, implying that new technologies will be required for the reliable detection of microproteins in fungi19. Despite the technological limitations faced by fungal researchers, several studies have showcased the function of microproteins across different growth conditions. In one investigation, researchers were able to identify the microprotein Nrs1 from a genome-wide overexpression screen in S. cerevisiae, whose upregulation rescued an otherwise inviable double gene-deletion mutant20. Nrs1 itself allows cells to overcome nitrogen-starved conditions and plays an important role in the regulatory circuitry involved in yeast budding20. Several other singular instances of microproteins serving as key players in regulatory pathways have been described, mostly from research on S. cerevisiae16. In another study in S. cerevisiae, researchers were able to identify 225 microproteins and plot how they are differentially- and sometimes, exclusively- expressed in response to UV stress, heat shock, and nutrient limitations, suggesting critical functions in different cellular adaptation contexts21.
There is therefore a strong implication that improved tools to investigate the fungal microproteome would result in numerous insights into stress response pathways and diverse aspects of fungal biology. While ribosome profiling and mass spectrometry-based proteomics may be adapted in other fungal species, these strategies could also be augmented by combining CRISPR screening and single-cell RNA sequencing to allow for the function of microproteins in fungi to be determined at scale, including the genome-wide effects of their perturbance on gene expression22. Similarly, improved models for the in silico prediction of microproteins that are not limited to one coding sequence per transcript, and that can predict sORFs and ORFs translated from non-AUG start codons, could now be applied to fungal species23. In addition, the optimization of new RNA-targeting CRISPR-based tools for fungi may allow for the discriminate targeting of sORF mRNA and characterization of microprotein function24.
Alternatively spliced proteins
Similar to microprotein formation, the capacity to include and exclude different sets of exons from a single gene through alternative splicing is also known to massively increase mRNA and protein diversity in eukaryotes25. Alternative splicing events have been demonstrated to play an important role in many facets of cell biology, including the establishment of drug resistance in human cancer cells and the pathogenesis of microbial parasites26,27. While alternative splicing is generally regarded as being less relevant in bacteria due to introns being either absent or very rare in prokaryotic species, splicing events are distinct in eukaryotes, where the number of genes that contain introns in fungi, for example, ranges massively from 4% to 99% across different species28. Despite this, alternative splicing was long seen as inconsequential to the cell in fungi, perceived as resulting in transcripts with either redundant or inoperative functionality29.
Recently, however, numerous mechanistic effects of alternative splicing have been established in fungi with impacts on growth, stress adaptation, infection, and immune recognition30. In the plant-parasitic fungus Shiraia bambusicola and the filamentous fungi Neurospora crassa and Aspergillus nidulans, alternative splicing has been observed to increase proteomic complexity and downstream functionality, particularly in response to different environmental stressors31,32,33. In the mushroom-forming Schizophyllum commune, alternative splicing was found to increase the total number of transcripts in the cell by 20%, and the majority of spliced transcripts were predicted to have alternative functions based on the fact that 70% of them had either lost or gained a functional domain compared to the non-spliced isoform29. In many yeast species, the rate of alternative splicing can be altered in response to stress, and further research into the molecular basis for the augmentation of genomic complexity from alternative splicing has demonstrated the capacity for dual localization of proteins encoded by the same gene to serve distinct functions in metabolism9,34,35. One study created a deletion set of all known introns in S. cerevisiae and identified the important roles introns have during competition and nutrient starvation36. Interestingly, in S. cerevisiae, once spliced out, introns themselves can become stably fixed in a cell and can act on different metabolic pathways, thus acting as regulatory ncRNAs37. In addition, expression via alternative transcriptional start sites and antisense transcription can be triggered in response to environmental cues in many fungi, including Metarhizium robertsii, A. nidulans, and Cryptococcus species, often resulting in changes in protein localization and downstream regulatory effects on gene expression38,39,40.
Alternative splicing may also be broadly associated with fungal pathogenicity, as it appears to be more prevalent in fungal pathogens, especially human fungal pathogens, than non-pathogenic taxa41. In the rice blast pathogen Magnaporthe oryzae, for example, deletion of the MoGrp1 protein involved in different splicing processes leads to a dramatic decrease in the virulence of the pathogen42. It has further been suggested that alternative splicing is potentially linked to drug susceptibility phenotypes in pathogenic fungi, and could even act as a target for the development of new therapeutics43. Indeed, alternative splicing seems to be differentially regulated specifically in response to certain antifungal drugs in N. crassa44. Another example involves the human pathogen Candida albicans with the oxidative stress-generating drug menadione45. In this study, researchers identified a case of differential drug resistance following deletion of the superoxide dismutase gene SOD3, where only overexpression of the spliced isoform of SOD3 rescued the mutant’s increased susceptibility to menadione45. As expected, menadione’s antifungal effect is partly based on its inhibition of the cell’s ability to perform alternative splicing45. While alternative splicing of introns clearly has underexplored and important roles in metabolism, antifungal drug resistance, and pathogenicity, the splicing of inteins — intervening sequences that are spliced out at the protein level — is also emerging as a process important to fungal biology and antifungal drug susceptibility. In the human pathogenic yeast Cryptococcus neoformans, different antifungal agents have been found to prevent the Prp8 intein from performing its essential role in cell viability and virulence via intein splicing, which has opened up new avenues for potential therapeutics against pathogenic fungi46,47.
Coinciding with the advent of long-read RNA sequencing technologies, new computational tools that allow for the sensitive detection of alternatively spliced transcripts have emerged in the past few years48. As these platforms continue to be improved upon, they may be adapted for the detection of mRNA isoforms in fungal species at a genome-wide level48. Further, adapting CRISPR-based platforms that allow for targeted deletion of single exons49, as well as multiplexed repression and activation of exons50, may allow fungal researchers to investigate the functional differences of spliced mRNA isoforms.
Non-coding RNAs
Non-coding RNAs (ncRNAs) include any RNA molecule in the cell that is generally not translated into a protein. While some classes of ncRNAs have long been acknowledged for playing crucial roles in the cell, including ribosomal RNA (rRNA) and transfer RNA (tRNA), the majority of ncRNA molecules were historically overlooked as being inert by-products of transcription51. However, more recent large-scale sequencing efforts have found that the majority of the genome can be transcribed, mostly into ncRNAs, and many divergent families of ncRNAs have been recognized for their unique and critical functions51. Among the many different classes of ncRNAs, regulatory RNAs include lncRNAs, miRNAs, and circRNAs. LncRNAs are ncRNAs longer than 200 nucleotides (nt), miRNAs have a length of around 19-25nt, and circRNAs differ in being non-linear and having a closed-loop structure. Each of these three types have different mechanisms of action, though in many cases exert epigenetic, RNA processing, and translational regulatory actions in the cell52,53,54. In human cells, all three of these regulatory ncRNA classes have been well studied in contributing to disease55,56. Despite their relatively simpler genomes, bacterial pathogens also utilize regulatory ncRNAs in diverse ways, some of which have been proposed as potential drug targets57. Regulatory ncRNAs, primarily lncRNAs, miRNAs, and circRNAs have all been identified in a myriad of disparate fungal taxa, though the extent to which these ncRNAs have been functionally characterized differ.
Long non-coding RNAs
Perhaps at the forefront of these inquiries are lncRNAs. In many fungi, lncRNA-encoding DNA can exist within and be transcribed from intergenic, intronic, sense, and antisense regions (Fig. 2)58. lncRNAs can be especially difficult to identify and characterize, as, unlike protein-coding genes, they seem to lack sequence conservation in mammalian cell lines, for example59. Despite this, lncRNAs have been described in many different fungal cellular processes including gene silencing and regulation, nutrient metabolism, histone modification, drug resistance, and virulence58,60. In fungi, lncRNAs have been most comprehensively studied in model yeasts. In S. cerevisiae, differential lncRNA abundance in distinct cell subpopulations has been suggested to play a regulatory role in cell and colony development61. Efforts have been made to characterize ncRNAs on a large scale in S. cerevisiae, where barcoded ncRNA deletion libraries have been screened to identify several intergenic ncRNAs essential to cell survival62. Another relatively large-scale approach taken in S. cerevisiae to explore lncRNA biology involved knocking out non-essential ORFs in combination with lncRNAs to generate double deletion mutant cells, allowing the characterization of lncRNAs via genetic interaction analysis63. This resulted in the identification of one lncRNA that acts in trans to regulate levels of distant telomeric single-stranded DNA, which is a necessary component of telomeric replication63. The researchers were, however, also able to implicate many other lncRNAs operating in a wide range of biological processes in the cell63. Indeed, there are several other well-defined cases of lncRNAs influencing the cell in yeast, including the specific regulatory roles that lncRNAs have on cell wall-related gene expression64. In the model fission yeast S. pombe, thousands of lncRNAs have been detected and have had their expressions monitored in response to different perturbations65. Amongst the lncRNAs in S. pombe, transcription of the well-characterized nc-tgp1 was found to play an important role in sensitivity to the drug thiabendazole as well as to hydroxyurea and caffeine by simply increasing nucleosome density, thereby preventing transcription of the neighboring tgp1 ORF (Fig. 2)66. lncRNA biology is also quickly expanding into other fungal taxa, including the yeast Pichia pastoris and the filamentous fungi N. crassa, where a vast amount of lncRNAs have been identified and assigned putative functions67,68.
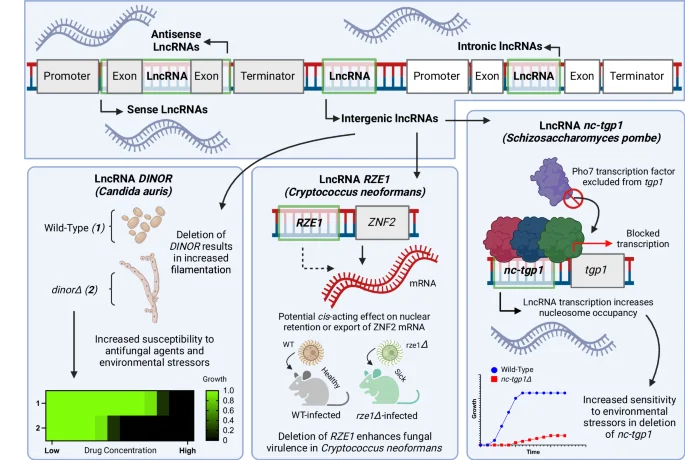
LncRNAs exist in several different orientations relative to canonical ORFs within fungal genomes, including in between exons (intronic), in between genes (intergenic), as well as within genes in either the same (sense) or opposite (antisense) direction as the larger ORF. Many lncRNAs have been characterized in diverse fungal species, including: the lncRNA DINOR in Candida auris that affects fungal morphology and drug resistance; the putatively cis-acting lncRNA sequence RZE1 in Cryptococcus neoformans that recapitulates virulence phenotypes of the neighboring gene ZNF2 when deleted and appears to affect the proportion of mRNA of ZNF2 that is localized to the nucleus; and the lncRNA nc-tgp1 that alters sensitivity to environmental stressors in Schizosaccharomyces pombe by increasing nucleosome occupancy when actively transcribed, excluding the transcription factor Pho7 from binding and activating expression of the downstream tgp1 gene. Figure 2, created with BioRender.com, released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license.
The field of fungal lncRNAs has also had a recent surge of interest in the context of pathogenic fungi. In many pathogenic Candida species, new bioinformatic and transcriptomic approaches continue to unveil more and more lncRNA transcripts in the cell69,70,71,72. Further, certain lncRNAs have been validated to exhibit significance in biological processes. One of the most striking examples involved a showcasing of the lncRNA named DINOR in the rapidly emerging multidrug-resistant human pathogen Candida auris, and its critical role in governing fungal pathogenicity, filamentous growth, and antifungal drug resistance (Fig. 2)73. DINOR was discovered via screening a genome-wide transposon mutagenesis library, therefore also inadvertently presenting a strong argument for constructing and screening more comprehensive mutant libraries that are not limited to canonical proteins73. On a larger scale, the differential expression of hundreds of lncRNAs cataloged across several important pathogenic Candida spp. during infection has also been characterized74.
The relevance of lncRNAs to virulence and metabolism is becoming clear in other crucial human pathogens as well, as a random insertional mutagenesis screen in C. neoformans uncovered RZE1, an lncRNA essential to pathogenicity via its predominately cis-acting regulatory function of the yeast-to-hypha transition (Fig. 2)75. In the pathogen Aspergillus sydowii, lncRNAs were found to play a role in tolerance to high NaCl stress, and in the related pathogen Aspergillus flavus, expression profiling of hundreds of lncRNAs suggested they are expressed and differentially localized in response to important environmental triggers, such as changes in temperature, osmotic stress, and CO276,77. In other fungal pathogens, including those that primarily infect and parasitize plants and insects, there has also been a rapid increase in interest in lncRNA function, where reports on the lncRNA landscape in Fusarium graminearum, Nosema ceranae, M. robertsii, and Cordyceps militaris, have all produced valuable preliminary insight into their functions in the cell78,79,80,81. Our understanding of lncRNAs in fungi and the diverse aspects of fungal biology they impact is continuing to expand.
MicroRNAs and micro-like RNAs
Research into other types of regulatory ncRNAs, namely miRNAs and circRNAs, has also shown promise for our improved understanding of fungal biology. RNA interference (RNAi) is a conserved mechanism in eukaryotes that typically involves small non-coding RNAs (sRNAs) ~ 19–24 nucleotides in length82. sRNAs work in concert with effector proteins to form an RNA-induced silencing complex (RISC) that can together silence mRNA translation via complementary binding to target RNA by the sRNA82. One category of sRNA in these systems is miRNA, which differs from other types due to their relatively indiscriminate binding patterns82. miRNAs also have many regulatory roles in the cell, rather than being simply a genome defense mechanism against invading viruses and transposons, as is the case for other sRNAs82. miRNAs were first discovered in Caenorhabditis elegans in 1993 and were initially considered to be absent in fungi until they were discovered in N. crassa in 201083,84. Since then, miRNAs and miRNA-like RNAs (milRNAs), which do not meet certain criteria established in other eukaryotic taxa to be included as miRNAs, have been identified in a considerable number of fungi, including Penicillium marneffei, Aspergillus fumigatus, Trichoderma reesei, Metarhizium anisopliae, Trichophyton rubrum, Sclerotinia sclerotiorum, C. albicans, and more83,85,86,87,88,89,90.
Much of the research into miRNAs and milRNAs in fungal species has involved employing sequencing approaches to validate their presence, and then analyzing their differential expression patterns in response to different growth conditions. Strategies such as these have allowed researchers to putatively classify miRNA and milRNA activity in important fungal cellular processes including thermal dimorphism, defense against mycoviral infection, cellulase production, mycelial growth and conidiogenesis, and sclerotial development, and also to predict their target RNAs in some cases83,85,86,87,88,89. Research in C. neoformans identified and profiled miRNAs, and found that miRNA sequences align to genomically-encoded transposons and pseudogenes, suggesting a role of miRNAs in regulating transposable element activity and the cryptic expression of pseudogenes91. Another phenomenon involves the spontaneous acquisition of drug resistance in the human fungal pathogen Mucor circinelloides via its RNAi-mediated gene silencing during growth in the presence of the antifungal drug tacrolimus92. Other studies have adopted a wider approach, such as in one case which involved employing computational tools to predict milRNAs and their RNA targets across 13 different plant fungal pathogen species93. This work identified several milRNA targets within the genome of their respective host plants, confirming a role for fungal milRNAs in suppressing plant host defense genes during infection93. Other examples of sRNA/milRNA-mediated silencing of host immunity by the plant pathogens Valsa mali and Botrytis cinerea have also been demonstrated and mechanistically characterized94,95. Despite these findings, it remains difficult to identify the activities of milRNAs in many cases, and it has been suggested that they may have other roles in genetic regulation besides mRNA cleaving, including translational repression or even DNA methylation96. Potential functions for sRNAs/miRNAs can also be overlooked, such as in one case where functional sRNA discovery in C. albicans was long impeded due to the widely used reference strain being uniquely deficient in a functional RNAi pathway, while the majority of other C. albicans isolates employ RNAi and sRNAs in the repression of telomere-associated genes90. As mi/milRNA annotations and their proposed functions in fungi continue to be assessed and improved upon97, the diverse roles of miRNAs and milRNAs across the fungal kingdom will become increasingly understood.
Circular RNAs
Circular RNAs (circRNAs) are the product of backsplicing, where the acceptor site of an upstream exon is towards its 5’ end, and the donor site of a downstream exon is towards its 3’ end, such that the exonic RNA folds in on itself and circularizes54. CircRNAs encode diverse cellular products, though they also seem to execute specific actions in the cell without being translated, including the inhibition of both miRNAs and the translation and activities of proteins54. It is also apparent that the ratio of circRNAs to linear RNA molecules is a tightly regulated process that has implications for disease and aging in humans54,98. CircRNAs of this kind were identified in fungi in 2014 in S. cerevisiae and S. pombe, but further research on them has been extremely limited98. In the past few years, sequencing efforts have led to hundreds or thousands of circRNAs being annotated in the genomes of the fungal pathogens M. oryzae, Ascosphaera apis, N. ceranae, and Ganoderma lucidum99,100,101,102. The ability of these circRNAs to act as “sponges” by competitively binding miRNAs was investigated and confirmed in all of these instances99,100,101,102. Interestingly, the expression patterns of different circRNAs often seemed to depend massively on the cell type or developmental stage of the fungus, though there has been a lack of characterization of individual circRNAs in specific cellular processes99,102,103. While intergenic, exonic, and intronic circRNAs exist, the proportion of circRNAs in either of the three groups seems to differ between fungal species101,103. Parallel efforts were also made in the human pathogenic T. rubrum, where researchers highlighted that one of the 4254 circRNAs they identified, Tru_circ07138_001, seemed to be highly conserved in ten other dermatophytic species analyzed, as well as in the distantly related red junglefowl Gallus gallus and C. elegans, indicating a shared role of this circRNA across the tree of life103.
The growing body of research on fungal regulatory ncRNAs serves as a compelling revelation of the importance of the hidden genome in fungi. New computational platforms, some of which leverage machine learning, that can identify regulatory ncRNAs from RNA sequencing data may be applied in fungi104,105. In addition, a diverse set of CRISPR-based screening platforms have already been demonstrated in human cells to study lncRNAs, miRNAs, and circRNAs en masse106,107,108,109. Indeed, harnessing new technologies developed for use in other species, along with continually improving techniques for functional genomic analysis in fungal taxa110,111, may greatly expand our understanding of these ncRNAs and their role in important facets of fungal biology.
Regulatory, repetitive, and canonically non-functional DNA
While many categories of ncRNAs and non-canonical proteins are beginning to be more clearly defined, there still exists parts of the genome that are more cryptic and underexplored. This involves the capacity of DNA to regulate gene expression independently of any transcribed or translated product, namely via non-coding regulatory elements (NCREs) such as promoters, silencers and enhancers, and insulators, that all contribute to cis-regulatory gene expression regulation112. However, it also includes regions of DNA that have historically been assumed to be non-functional, including repetitive regions and transposons, as well as pseudogenes and dubious ORFs, much of which has often been referred to as ‘junk’ DNA. ‘Junk’ DNA, has traditionally referred to any DNA sequence that does not play a known role in any cellular process, and has long been assumed to represent the vast majority of the DNA in the genome112,113. However, what constituent non-coding DNA is actually non-functional remains a contested topic, especially when considering the C-value paradox, which involves the discrepancy between the expectation that more complex organisms should tend to have larger genomes, and the reality that they often do not114. While the Encyclopedia of DNA Elements (ENCODE) project proposed that, in fact, 80% of the human genome is linked to biochemically active processes, criticisms of this claim involve the fact that their definition of biochemical activity does not extend to what is actually functional in the human cell, and that much of the DNA in a given genome remains to be assigned a true function114,115. Regardless, there are still many clear and validated examples of regions of ‘junk’ DNA contributing to diverse cellular phenomena113. These include pseudogenes, highly repetitive DNA, and transposable elements113. These groups of DNA have already been shown to be potential regulators of protein-coding DNA in some eukaryotes, as well as been demonstrated as effective therapeutic targets in cancer cells, but they remain relatively poorly studied in fungi116,117.
Non-coding regulatory elements
The identification and precise mapping of NCREs varies widely between different fungal taxa and often focuses on trans-acting regulatory components like transcription factors118,119. However, there have been important discoveries made on the topic of NCREs in fungi, and many efforts have been made, particularly in recent years, to employ methods that would allow for the mapping and functional analysis of these cis-regulatory components in Saccharomyces species120,121,122. Related phenomena have been identified in S. cerevisiae, in particular, where the genomic regions that encode tRNA seem to act as chromatin insulators with strong implications in preventing gene repression and activation123,124. Systematically categorizing fungal genetic circuits and NCREs may help uncover novel regulatory mechanisms of fungal pathogenesis and allow researchers to better harness fungal metabolism for industrial applications and for the production of important metabolites. Attempting to characterize cis-regulatory DNA is difficult, in part due to the complex nature of the interactions between transcription factors and a dynamic 3D DNA landscape125. However, CRISPR screens hold promise for functionally characterizing NCREs at scale126. In addition, the combination of improved machine learning models with new technologies that can produce long synthetic DNA molecules may help us to investigate transcription factor binding in the context of a locus large enough to account for high-level chromatin architecture and DNA-DNA interactions125.
Transposable elements and repetitive DNA
Another underexplored component of the fungal genome involves transposable elements (TEs). TEs encompass an array of different DNA sequences, some of which possess genes that enable them to change position in the genome, causing disruptions in gene function or alterations of local gene expression, and in some cases transport ‘cargo’ genes that are not required for propagation of the TE and may be of benefit to the host127,128. Additionally, the repetitive nature of non-mobile TE sequences scattered throughout the genome can facilitate structural rearrangements and shape genomic architecture127. The proportion of the genome that is made up of TEs varies from ~0% to upwards of 30% in fungi127. While the extent of their regulatory roles in shaping fungal phenotypes remains contested, they are linked to the expression of genes and their evolution in fungi129. Indeed, in M. oryzae, the presence of TEs appears to increase genetic diversity in neighboring genes, which in turn drives host specialization of the pathogen130. Studies done on the fungal plant pathogen Zymoseptoria tritici have revealed that TE insertions can directly regulate melanin biosynthesis, and therefore fungal virulence, as well as multi-drug resistance via fungicide efflux upregulation131,132. TE insertions also drive adaptation during host infection and contribute to the acquisition of drug resistance in Cryptococcus (Fig. 3)133,134. Transposon mobility in these cases displays a temperature-dependent pattern, underscoring their influence on the cell’s ability to respond to environmental changes (Fig. 3)133,135. Bioinformatic-based findings in other fungal pathogens also suggest that genes under TE influence are often repressed and that there tends to be a correlation between TE prevalence in the genome and symbiotic tendencies127,136.
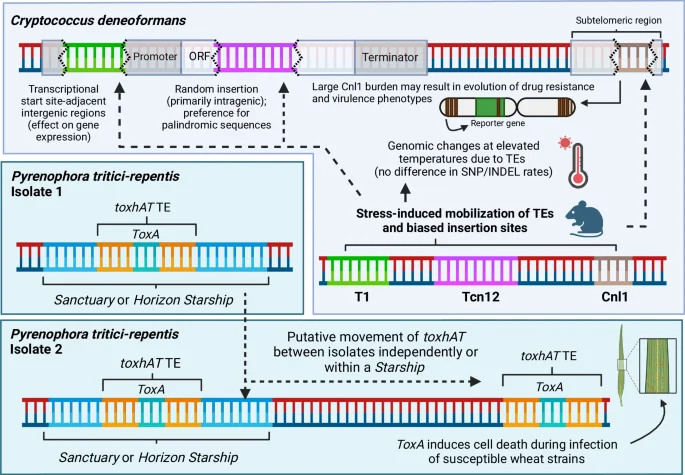
In the human fungal pathogen Cryptococcus deneoformans, the mobility of several different TEs is stress-responsive, including during infection and at the elevated (host-relevant) temperature of 37 °C. Insertion of mobilized TEs in this pathogen also appears to be biased depending on the type of TE, and has been associated with a decrease in susceptibility to antifungal drugs. The accumulation of the Cnl1 TE at subtelomeric regions, for example, can result in additional copies of Cnl1 being driven towards other sites in the genome, which may influence drug response and virulence phenotypes. The rate of other genomic changes, including from single nucleotide polymorphisms (SNPs) and insertion/deletion (indel) mutations, remain unchanged at elevated temperatures, suggesting TE mobility is a primary driver of genomic change during heat stress. In the plant fungal pathogen Pyrenophora tritici-repentis, the toxhAT TE contains the ToxA gene which can induce cell death in susceptible wheat strains during infection. The toxhAT TE has been observed to move between isolates of the same species, as well as between different species entirely. While toxhAT has been observed to exist within larger Starship TEs, there is also evidence to suggest that it is able to mobilize from the genome independently. Figure 3, created with BioRender.com, released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license.
A role for fungal TEs in horizontal gene transfer has also recently emerged based on the identification of very large transposons named Starships, which can be hundreds of kilobases in size and carry genes important to fungal survival and adaptation137. Many different Starships have been identified with high sequence similarity in divergent fungal taxa, suggesting they are able to migrate between species independently of the fungal host’s cellular machinery138,139,140. For example, the Hephaestus Starship locus that confers heavy metal resistance to the emerging infectious mould Paecilomyces variotii appears to have been shared with the related species Paecilomyces lecythidis, as well as Penicillium fuscoglaucum138,141. The nested nature of these giant TEs also implies that they are highly diverse. The ToxA gene that confers virulence to fungal wheat pathogens has been transferred horizontally between several species, and appears to exist within a ~14 kb TE named ToxhAT (Fig. 3)128. However, the ToxhAT locus itself has been identified within two larger Starship elements named Horizon and Sanctuary (Fig. 3)142,143. Considering TEs and larger Starships may be involved in the regulation and active exchange of genes involved in stress adaptation and pathogenicity within and between fungal taxa, further research being done to mine fungal genomes for their presence will be imperative in functionally characterizing these genetic factors across the fungal kingdom144.
Analyzing large repetitive regions of DNA as a whole, some of which include both non-coding DNA and ORFs, can also be a strategy for broader genome characterization. In an attempt to understand the plastic and rapidly adapting nature of the C. albicans genome, one study found that all segmental aneuploidy events — which are a critical part of C. albicans pathogenesis and drug resistance145 — occurred at long repeat sequences (anywhere from 65–6499bp in length)146. Here, these long repeat regions of DNA seemed to be necessary for many of the adaptive traits that C. albicans can harness to survive in a diverse set of challenging environments146. This is not entirely surprising as it has been long understood that large repeat regions of this kind seem to be relatively more amenable to rapid evolution in filamentous fungal plant pathogens, and tend to harbor virulence genes147. Indeed, new telomere-to-telomere sequencing platforms that can accurately annotate highly repetitive DNA that have already been applied in some fungi and oomycetes is a promising avenue for accurately identifying transposons and functional repeat elements at a genome-wide level148,149.
Pseudogenes and Dubious ORFs
Another component of ‘junk’ DNA is pseudogenes. Pseudogenes are genes that share close sequence similarities with canonical ORFs but have a disruptive mutation that precludes their transcription or leads to products with either reduced or abolished function150. For over a decade, the idea of pseudogenes encoding important regulatory products and having divergent impacts on cellular fitness has been demonstrated, but this research has been predominantly focused on human cells116. However, pseudogenes have been identified in many different fungal species, and have been a valuable means of understanding evolution and loss of pathogenicity150,151,152. A landmark study in S. cerevisiae from over two decades ago found active expression of some pseudogenes and posited that some may have impacts on the fungal stress rseponse153. Despite the advancements being made in identifying functional pseudogenes, there remains much to be characterized about the impacts of pseudogenes in most fungal species153. However, comparative analysis of pseudogene profiles between closely related species may be used to help identify genes responsible for any divergent functional capacities between the species151. Further, methods for applying CRISPR-based tools to study pseudogenes have been outlined and could be applied in fungal species154.
While not ‘junk’ DNA per se, dubious ORFs also represent an interesting area of functional genomic research. Dubious ORFs are ORFs that were originally suspected to not encode for a functional product, often based on the criteria that they are not conserved in any related species from the same taxa and that there is no experimental evidence of a resulting gene product155. ORFs labeled as dubious also tend to overlap with microproteins and sORFs, since both have traditionally been considered to not generate anything useful to the cell, even if some are known to still be translated155,156. Despite this categorization, an evaluative study on the accuracy of dubious ORF classifications in S. cerevisiae showed that many of these ORFs, indeed, produced detectable transcripts and/or products of translation157. The implications of this study have become clear with the emergence of a few notable examples in S. cerevisiae where the specific deletion of dubious ORFs resulted in prominent phenotypes associated with protein burden regulation and mitochondrial DNA maintenance158,159. This notion may also extend into other fungal taxa as it was recently shown in the human pathogen C. albicans that many previously labeled dubious ORFs are actively transcribed and translated, and that the rates of which seem to be differentially regulated during C. albicans’ morphological transitions156.
It has also been demonstrated that non-coding DNA is positively selected for in certain fungi, and does not always represent genetic relics with simple coincidental influence on gene expression. One study done on the fungal plant pathogen genus Colletotrichum illustrated many instances of positive selection for non-coding DNA during infection, which they suggest implies a regulatory role of this non-coding DNA160. It has elsewhere been proposed that the persistence of non-coding DNA in fungi is due to its function in harboring regulatory ncRNAs, or its role in intragenic DNA methylation, like in other eukaryotes161,162.
Conclusions and future perspectives
Many facets of fungal biology remain underexplored. However, the recent discovery of novel components of the fungal hidden genome may help us develop a more holistic understanding of fungal genetics and genomics. New technologies, particularly CRISPR-based techniques, have proven to be powerful tools to characterize these hidden components of the genome in non-fungal cell systems8. Harnessing new platforms developed for functional genomics in fungi may similarly help characterize important processes regulated by the non-coding genome and products of non-canonical expression. Numerous iterations of CRISPR technologies with disparate mechanisms of action have been demonstrated in diverse fungal taxa, including those that may be amenable to specific interrogation of hidden genome components that are nested within larger canonical sequences via differential expression163,164,165, those with a sensitive resolution for editing very small sequences of DNA166, and those that can discriminately target RNA molecules167. Thus, as more of these technologies are applied in fungi, a more robust characterization of non-canonical fungal genomes will emerge.
References
-
Adhikari, S. et al. A high-stringency blueprint of the human proteome. Nat. Commun. 11, 5301 (2020).
Google Scholar
-
Wright, B. W., Yi, Z., Weissman, J. S. & Chen, J. The dark proteome: translation from noncanonical open reading frames. Trends Cell Biol. 32, 243–258 (2022).
Google Scholar
-
Ruiz Cuevas, M. V. et al. Most non-canonical proteins uniquely populate the proteome or immunopeptidome. Cell Rep. 34, 108815 (2021).
Google Scholar
-
Prensner, J. R. et al. Noncanonical open reading frames encode functional proteins essential for cancer cell survival. Nat. Biotechnol. 39, 697–704 (2021).
Google Scholar
-
Orr, M. W., Mao, Y., Storz, G. & Qian, S.-B. Alternative ORFs and small ORFs: shedding light on the dark proteome. Nucleic Acids Res. 48, 1029–1042 (2020).
Google Scholar
-
Lu, S. et al. A hidden human proteome encoded by ‘non-coding’ genes. Nucleic Acids Res. 47, 8111–8125 (2019).
Google Scholar
-
Statello, L., Guo, C.-J., Chen, L.-L. & Huarte, M. Author Correction: Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 22, 159 (2021).
Google Scholar
-
Montalbano, A., Canver, M. C. & Sanjana, N. E. High-throughput approaches to pinpoint function within the noncoding genome. Mol. Cell 68, 44–59 (2017).
Google Scholar
-
Sieber, P. et al. Comparative study on alternative splicing in human fungal pathogens suggests its involvement during host invasion. Front. Microbiol. 9, 2313 (2018).
Google Scholar
-
Balarezo-Cisneros, L. N. et al. Functional and transcriptional profiling of non-coding RNAs in yeast reveal context-dependent phenotypes and in trans effects on the protein regulatory network. PLoS Genet 17, e1008761 (2021).
Google Scholar
-
Huberman, L. B. Developing functional genomics platforms for fungi. mSystems 6, e0073021 (2021).
Google Scholar
-
Hassel, K. R., Brito-Estrada, O. & Makarewich, C. A. Microproteins: overlooked regulators of physiology and disease. iScience 26, 106781 (2023).
Google Scholar
-
Martinez, T. F. et al. Accurate annotation of human protein-coding small open reading frames. Nat. Chem. Biol. 16, 458–468 (2020).
Google Scholar
-
Chu, Q. et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat. Commun. 10, 4883 (2019).
Google Scholar
-
Garai, P. & Blanc-Potard, A. Uncovering small membrane proteins in pathogenic bacteria: Regulatory functions and therapeutic potential. Mol. Microbiol. 114, 710–720 (2020).
Google Scholar
-
Erpf, P. E. & Fraser, J. A. The long history of the diverse roles of short ORFs: sPEPs in fungi. Proteomics 18, e1700219 (2018).
Google Scholar
-
Mat-Sharani, S. & Firdaus-Raih, M. Computational discovery and annotation of conserved small open reading frames in fungal genomes. BMC Bioinforma. 19, 551 (2019).
Google Scholar
-
Yang, H., Li, Q., Stroup, E. K., Wang, S. & Ji, Z. Widespread stable noncanonical peptides identified by integrated analyses of ribosome profiling and ORF features. Nat. Commun. 15, 1932 (2024).
Google Scholar
-
Huraiova, B. et al. Proteomic analysis of meiosis and characterization of novel short open reading frames in the fission yeast Schizosaccharomyces pombe. Cell Cycle 19, 1777–1785 (2020).
Google Scholar
-
Tollis, S. et al. The microprotein Nrs1 rewires the G1/S transcriptional machinery during nitrogen limitation in budding yeast. PLoS Biol. 20, e3001548 (2022).
Google Scholar
-
Sun, Y., Huang, J., Wang, Z., Pan, N. & Wan, C. Identification of microproteins in Saccharomyces cerevisiae under different stress conditions. J. Proteome Res. 21, 1939–1947 (2022).
Google Scholar
-
Chen, J. et al. Pervasive functional translation of noncanonical human open reading frames. Science 367, 1140–1146 (2020).
Google Scholar
-
Valdivia-Francia, F. & Sendoel, A. No country for old methods: new tools for studying microproteins. iScience 27, 108972 (2024).
Google Scholar
-
Treichel, A. J. & Bazzini, A. A. Casting CRISPR-Cas13d to fish for microprotein functions in animal development. iScience 25, 105547 (2022).
Google Scholar
-
Nilsen, T. W. & Graveley, B. R. Expansion of the eukaryotic proteome by alternative splicing. Nature 463, 457–463 (2010).
Google Scholar
-
Yeoh, L. M. et al. Alternative splicing is required for stage differentiation in malaria parasites. Genome Biol. 20, 151 (2019).
Google Scholar
-
Melnyk, J. E. et al. Targeting a splicing-mediated drug resistance mechanism in prostate cancer by inhibiting transcriptional regulation by PKCβ1. Oncogene 41, 1536–1549 (2022).
Google Scholar
-
Muzafar, S., Sharma, R. D., Chauhan, N. & Prasad, R. Intron distribution and emerging role of alternative splicing in fungi. FEMS Microbiol. Lett. 368, fnab135 (2021).
Google Scholar
-
Gehrmann, T. et al. Schizophyllum commune has an extensive and functional alternative splicing repertoire. Sci. Rep. 6, 33640 (2016).
Google Scholar
-
Fang, S. et al. The occurrence and function of alternative splicing in fungi. Fungal Biol. Rev. 34, 178–188 (2020).
Google Scholar
-
Liu, X.-Y., Fan, L., Gao, J., Shen, X.-Y. & Hou, C.-L. Global identification of alternative splicing in Shiraia bambusicola and analysis of its regulation in hypocrellin biosynthesis. Appl. Microbiol. Biotechnol. 104, 211–223 (2020).
Google Scholar
-
Leal, J. et al. A splice variant of the Neurospora crassa hex-1 transcript, which encodes the major protein of the Woronin body, is modulated by extracellular phosphate and pH changes. FEBS Lett. 583, 180–184 (2009).
Google Scholar
-
Trevisan, G. L. et al. Transcription of Aspergillus nidulans pacC is modulated by alternative RNA splicing of palB. FEBS Lett. 585, 3442–3445 (2011).
Google Scholar
-
Strijbis, K., van den Burg, J., Visser, W. F., van den Berg, M. & Distel, B. Alternative splicing directs dual localization of Candida albicans 6-phosphogluconate dehydrogenase to cytosol and peroxisomes. FEMS Yeast Res. 12, 61–68 (2012).
Google Scholar
-
Juneau, K., Nislow, C. & Davis, R. W. Alternative splicing of PTC7 in Saccharomyces cerevisiae determines protein localization. Genetics 183, 185–194 (2009).
Google Scholar
-
Parenteau, J. et al. Introns are mediators of cell response to starvation. Nature 565, 612–617 (2019).
Google Scholar
-
Morgan, J. T., Fink, G. R. & Bartel, D. P. Excised linear introns regulate growth in yeast. Nature 565, 606–611 (2019).
Google Scholar
-
Guo, N. et al. Alternative transcription start site selection in Mr-OPY2 controls lifestyle transitions in the fungus Metarhizium robertsii. Nat. Commun. 8, 1565 (2017).
Google Scholar
-
Sibthorp, C. et al. Transcriptome analysis of the filamentous fungus Aspergillus nidulans directed to the global identification of promoters. BMC Genomics 14, 847 (2013).
Google Scholar
-
Dang, T. T. V. et al. Alternative TSS use is widespread in Cryptococcus fungi in response to environmental cues and regulated genome-wide by the transcription factor Tur1. bioRxiv https://doi.org/10.1101/2023.07.18.549460 (2024). 2023.07.18.549460.
Google Scholar
-
Grützmann, K. et al. Fungal alternative splicing is associated with multicellular complexity and virulence: a genome-wide multi-species study. DNA Res. 21, 27–39 (2014).
Google Scholar
-
Gao, X. et al. A glycine-rich protein MoGrp1 functions as a novel splicing factor to regulate fungal virulence and growth in Magnaporthe oryzae. Phytopathol. Res. 1, 1–15 (2019).
Google Scholar
-
Jayaguru, P. & Raghunathan, M. Group I intron renders differential susceptibility of Candida albicans to Bleomycin. Mol. Biol. Rep. 34, 11–17 (2007).
Google Scholar
-
Mendes, N. S., Silva, P. M., Silva-Rocha, R., Martinez-Rossi, N. M. & Rossi, A. Pre-mRNA splicing is modulated by antifungal drugs in the filamentous fungus Neurospora crassa. FEBS Open Bio 6, 358–368 (2016).
Google Scholar
-
Muzafar, S. et al. Identification of genomewide alternative splicing events in sequential, isogenic clinical isolates of Candida albicans reveals a novel mechanism of drug resistance and tolerance to cellular stresses. mSphere 5, e00608–e00620 (2020).
Google Scholar
-
Tharappel, A. M. et al. Calcimycin inhibits Cryptococcus neoformans in vitro and in vivo by targeting the Prp8 intein splicing. ACS Infect. Dis. 8, 1851–1868 (2022).
Google Scholar
-
Li, Z. et al. Small-molecule inhibitors for the Prp8 intein as antifungal agents. Proc. Natl. Acad. Sci. USA 118, e2008815118 (2021).
Google Scholar
-
Su, Y. et al. Comprehensive assessment of mRNA isoform detection methods for long-read sequencing data. Nat. Commun. 15, 3972 (2024).
Google Scholar
-
Xiao, M.-S. et al. Genome-scale exon perturbation screens uncover exons critical for cell fitness. Mol. Cell 84, 2553–2572.e19 (2024).
Google Scholar
-
Li, J. D., Taipale, M. & Blencowe, B. J. Efficient, specific, and combinatorial control of endogenous exon splicing with dCasRx-RBM25. Mol. Cell 84, 2573–2589.e5 (2024).
Google Scholar
-
Zhang, P., Wu, W., Chen, Q. & Chen, M. Non-coding RNAs and their integrated networks. J. Integr. Bioinform. 16, 20190027 (2019).
Google Scholar
-
Mattick, J. S. et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 24, 430–447 (2023).
Google Scholar
-
Shang, R., Lee, S., Senavirathne, G. & Lai, E. C. microRNAs in action: biogenesis, function and regulation. Nat. Rev. Genet. 24, 816–833 (2023).
Google Scholar
-
Kristensen, L. S. et al. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 20, 675–691 (2019).
Google Scholar
-
Yang, Y. et al. The roles of miRNA, lncRNA and circRNA in the development of osteoporosis. Biol. Res. 53, 40 (2020).
Google Scholar
-
Li, C. et al. Crosstalk of mRNA, miRNA, lncRNA, and circRNA and their regulatory pattern in pulmonary fibrosis. Mol. Ther. Nucleic Acids 18, 204–218 (2019).
Google Scholar
-
Eichner, H., Karlsson, J. & Loh, E. The emerging role of bacterial regulatory RNAs in disease. Trends Microbiol. 30, 959–972 (2022).
Google Scholar
-
Li, J., Liu, X., Yin, Z., Hu, Z. & Zhang, K.-Q. An overview on identification and regulatory mechanisms of long non-coding RNAs in fungi. Front. Microbiol. 12, 638617 (2021).
Google Scholar
-
Johnsson, P., Lipovich, L., Grandér, D. & Morris, K. V. Evolutionary conservation of long non-coding RNAs; sequence, structure, function. Biochim. Biophys. Acta 1840, 1063–1071 (2014).
Google Scholar
-
Dhingra, S. Role of non-coding RNAs in fungal pathogenesis and antifungal drug responses. Curr. Clin. Microbiol. Rep. 7, 133–141 (2020).
Google Scholar
-
Wilkinson, D. et al. Long noncoding RNAs in yeast cells and differentiated subpopulations of yeast colonies and biofilms. Oxid. Med. Cell. Longev. 2018, 4950591 (2018).
Google Scholar
-
Parker, S. et al. Large-scale profiling of noncoding RNA function in yeast. PLoS Genet 14, e1007253 (2018).
Google Scholar
-
Kyriakou, D. et al. Functional characterisation of long intergenic non-coding RNAs through genetic interaction profiling in Saccharomyces cerevisiae. BMC Biol. 14, 106 (2016).
Google Scholar
-
Novačić, A., Vučenović, I., Primig, M. & Stuparević, I. Non-coding RNAs as cell wall regulators in Saccharomyces cerevisiae. Crit. Rev. Microbiol. 46, 15–25 (2020).
Google Scholar
-
Atkinson, S. R. et al. Long noncoding RNA repertoire and targeting by nuclear exosome, cytoplasmic exonuclease, and RNAi in fission yeast. RNA 24, 1195–1213 (2018).
Google Scholar
-
Ard, R., Tong, P. & Allshire, R. C. Long non-coding RNA-mediated transcriptional interference of a permease gene confers drug tolerance in fission yeast. Nat. Commun. 5, 5576 (2014).
Google Scholar
-
Sun, W.-H., Wang, Y.-Z., Xu, Y. & Yu, X.-W. Genome-wide analysis of long non-coding RNAs in Pichia pastoris during stress by RNA sequencing. Genomics 111, 398–406 (2019).
Google Scholar
-
Cemel, I. A., Ha, N., Schermann, G., Yonekawa, S. & Brunner, M. The coding and noncoding transcriptome of Neurospora crassa. BMC Genomics 18, 978 (2017).
Google Scholar
-
Linde, J. et al. Defining the transcriptomic landscape of Candida glabrata by RNA-Seq. Nucleic Acids Res. 43, 1392–1406 (2015).
Google Scholar
-
Donovan, P. D., Schröder, M. S., Higgins, D. G. & Butler, G. Identification of non-coding RNAs in the Candida parapsilosis species group. PLoS One 11, e0163235 (2016).
Google Scholar
-
Sellam, A. et al. Experimental annotation of the human pathogen Candida albicans coding and noncoding transcribed regions using high-resolution tiling arrays. Genome Biol. 11, R71 (2010).
Google Scholar
-
Mathur, K., Singh, B., Puria, R. & Nain, V. In silico genome wide identification of long non-coding RNAs differentially expressed during Candida auris host pathogenesis. Arch. Microbiol. 206, 253 (2024).
Google Scholar
-
Gao, J. et al. LncRNA DINOR is a virulence factor and global regulator of stress responses in Candida auris. Nat. Microbiol 6, 842–851 (2021).
Google Scholar
-
Hovhannisyan, H. & Gabaldón, T. The long non-coding RNA landscape of Candida yeast pathogens. Nat. Commun. 12, 7317 (2021).
Google Scholar
-
Chacko, N. et al. The lncRNA RZE1 controls cryptococcal morphological transition. PLoS Genet 11, e1005692 (2015).
Google Scholar
-
Jiménez-Gómez, I. et al. Surviving in the brine: a multi-omics approach for understanding the physiology of the halophile fungus Aspergillus sydowii at saturated NaCl concentration. Front. Microbiol. 13, 840408 (2022).
Google Scholar
-
Davati, N. & Ghorbani, A. Discovery of long non-coding RNAs in Aspergillus flavus response to water activity, CO2 concentration, and temperature changes. Sci. Rep. 13, 10330 (2023).
Google Scholar
-
Kim, W., Miguel-Rojas, C., Wang, J., Townsend, J. P. & Trail, F. Developmental dynamics of long noncoding RNA expression during sexual fruiting body formation in Fusarium graminearum. MBio 9, e01292–1 (2018).
Google Scholar
-
Wang, Z., Jiang, Y., Wu, H., Xie, X. & Huang, B. Genome-wide identification and functional prediction of long non-coding RNAs involved in the heat stress response in Metarhizium robertsii. Front. Microbiol. 10, 2336 (2019).
Google Scholar
-
Guo, R. et al. First identification of long non-coding RNAs in fungal parasite Nosema ceranae. Apidologie 49, 660–670 (2018).
Google Scholar
-
Wang, Y. et al. XRN1-associated long non-coding RNAs may contribute to fungal virulence and sexual development in entomopathogenic fungus Cordyceps militaris. Pest Manag. Sci. 75, 3302–3311 (2019).
Google Scholar
-
Dang, Y., Yang, Q., Xue, Z. & Liu, Y. RNA interference in fungi: pathways, functions, and applications. Eukaryot. Cell 10, 1148–1155 (2011).
Google Scholar
-
Zhou, J. et al. Identification of microRNA-like RNAs in a plant pathogenic fungus Sclerotinia sclerotiorum by high-throughput sequencing. Mol. Genet. Genomics 287, 275–282 (2012).
Google Scholar
-
Lee, H.-C. et al. Diverse pathways generate microRNA-like RNAs and Dicer-independent small interfering RNAs in fungi. Mol. Cell 38, 803–814 (2010).
Google Scholar
-
Lau, S. K. P. et al. Identification of microRNA-like RNAs in mycelial and yeast phases of the thermal dimorphic fungus Penicillium marneffei. PLoS Negl. Trop. Dis. 7, e2398 (2013).
Google Scholar
-
Özkan, S., Mohorianu, I., Xu, P., Dalmay, T. & Coutts, R. H. A. Profile and functional analysis of small RNAs derived from Aspergillus fumigatus infected with double-stranded RNA mycoviruses. BMC Genomics 18, 416 (2017).
Google Scholar
-
Kang, K. et al. Identification of microRNA-Like RNAs in the filamentous fungus Trichoderma reesei by solexa sequencing. PLoS One 8, e76288 (2013).
Google Scholar
-
Zhou, Q., Wang, Z., Zhang, J., Meng, H. & Huang, B. Genome-wide identification and profiling of microRNA-like RNAs from Metarhizium anisopliae during development. Fungal Biol. 116, 1156–1162 (2012).
Google Scholar
-
Wang, L. et al. Integrated microRNA and mRNA analysis in the pathogenic filamentous fungus Trichophyton rubrum. BMC Genomics 19, 933 (2018).
Google Scholar
-
Iracane, E. et al. Identification of an active RNAi pathway in Candida albicans. Proc. Natl Acad. Sci. USA 121, e2315926121 (2024).
Google Scholar
-
Jiang, N., Yang, Y., Janbon, G., Pan, J. & Zhu, X. Identification and functional demonstration of miRNAs in the fungus Cryptococcus neoformans. PLoS One 7, e52734 (2012).
Google Scholar
-
Calo, S. et al. Antifungal drug resistance evoked via RNAi-dependent epimutations. Nature 513, 555–558 (2014).
Google Scholar
-
Mathur, M., Nair, A. & Kadoo, N. Plant-pathogen interactions: microRNA-mediated trans-kingdom gene regulation in fungi and their host plants. Genomics 112, 3021–3035 (2020).
Google Scholar
-
Xu, M. et al. A fungal microRNA-like RNA subverts host immunity and facilitates pathogen infection by silencing two host receptor-like kinase genes. N. Phytol. 233, 2503–2519 (2022).
Google Scholar
-
He, B. et al. Fungal small RNAs ride in extracellular vesicles to enter plant cells through clathrin-mediated endocytosis. Nat. Commun. 14, 4383 (2023).
Google Scholar
-
Chen, R. et al. Exploring microRNA-like small RNAs in the filamentous fungus Fusarium oxysporum. PLoS One 9, e104956 (2014).
Google Scholar
-
Johnson, N. R., Larrondo, L. F., Álvarez, J. M. & Vidal, E. A. Comprehensive re-analysis of hairpin small RNAs in fungi reveals loci with conserved links. Elife 11, e83691 (2022).
Google Scholar
-
Wang, P. L. et al. Circular RNA is expressed across the eukaryotic tree of life. PLoS One 9, e90859 (2014).
Google Scholar
-
Yuan, J., Wang, Z., Xing, J., Yang, Q. & Chen, X.-L. Genome-wide Identification and characterization of circular RNAs in the rice blast fungus Magnaporthe oryzae. Sci. Rep. 8, 6757 (2018).
Google Scholar
-
Guo, R. et al. Systematic investigation of circular RNAs in Ascosphaera apis, a fungal pathogen of honeybee larvae. Gene 678, 17–22 (2018).
Google Scholar
-
Guo, R. et al. Genome-wide identification of circular RNAs in fungal parasite Nosema ceranae. Curr. Microbiol. 75, 1655–1660 (2018).
Google Scholar
-
Shao, J. et al. Identification and characterization of circular RNAs in Ganoderma lucidum. Sci. Rep. 9, 16522 (2019).
Google Scholar
-
Cao, X. et al. Genome-wide identification and functional analysis of circRNAs in Trichophyton rubrum conidial and mycelial stages. BMC Genomics 23, 21 (2022).
Google Scholar
-
Vromman, M. et al. Large-scale benchmarking of circRNA detection tools reveals large differences in sensitivity but not in precision. Nat. Methods 20, 1159–1169 (2023).
Google Scholar
-
Li, M. & Liang, C. LncDC: a machine learning-based tool for long non-coding RNA detection from RNA-Seq data. Sci. Rep. 12, 19083 (2022).
Google Scholar
-
Bester, A. C. et al. An integrated genome-wide CRISPRa approach to functionalize lncRNAs in drug resistance. Cell 173, 649–664.e20 (2018).
Google Scholar
-
Wallace, J. et al. Genome-wide CRISPR-Cas9 screen identifies MicroRNAs that regulate myeloid leukemia cell growth. PLoS One 11, e0153689 (2016).
Google Scholar
-
Li, S. et al. Screening for functional circular RNAs using the CRISPR-Cas13 system. Nat. Methods 18, 51–59 (2021).
Google Scholar
-
Montero, J. J. et al. Genome-scale pan-cancer interrogation of lncRNA dependencies using CasRx. Nat. Methods 21, 584–596 (2024).
Google Scholar
-
Uthayakumar, D., Sharma, J., Wensing, L. & Shapiro, R. S. CRISPR-based genetic manipulation of Candida species: historical perspectives and current approaches. Front Genome Ed. 2, 606281 (2020).
Google Scholar
-
Ross, R. L. & Santiago-Tirado, F. H. Advanced genetic techniques in fungal pathogen research. mSphere 9, e0064323 (2024).
Google Scholar
-
Pang, B., van Weerd, J. H., Hamoen, F. L. & Snyder, M. P. Identification of non-coding silencer elements and their regulation of gene expression. Nat. Rev. Mol. Cell Biol. 24, 383–395 (2023).
Google Scholar
-
Palazzo, A. F. & Gregory, T. R. The case for junk DNA. PLoS Genet 10, e1004351 (2014).
Google Scholar
-
Eddy, S. R. The C-value paradox, junk DNA and ENCODE. Curr. Biol. 22, R898–R899 (2012).
Google Scholar
-
Ecker, J. R. et al. Genomics: ENCODE explained. Nature 489, 52–55 (2012).
Google Scholar
-
Pink, R. C. et al. Pseudogenes: pseudo-functional or key regulators in health and disease? RNA 17, 792–798 (2011).
Google Scholar
-
Brocks, D. et al. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat. Genet. 49, 1052–1060 (2017).
Google Scholar
-
Hughes, T. R. & de Boer, C. G. Mapping yeast transcriptional networks. Genetics 195, 9–36 (2013).
Google Scholar
-
Daguerre, Y. et al. Regulatory networks underlying mycorrhizal development delineated by genome-wide expression profiling and functional analysis of the transcription factor repertoire of the plant symbiotic fungus Laccaria bicolor. BMC Genomics 18, 737 (2017).
Google Scholar
-
Kita, R., Venkataram, S., Zhou, Y. & Fraser, H. B. High-resolution mapping of cis-regulatory variation in budding yeast. Proc. Natl. Acad. Sci. USA 114, E10736–E10744 (2017).
Google Scholar
-
Renganaath, K. et al. Systematic identification of cis-regulatory variants that cause gene expression differences in a yeast cross. Elife 9, e62669 (2020).
Google Scholar
-
Shih, C.-H. & Fay, J. Cis-regulatory variants affect gene expression dynamics in yeast. Elife 10, e68469 (2021).
Google Scholar
-
Simms, T. A. et al. TFIIIC binding sites function as both heterochromatin barriers and chromatin insulators in Saccharomyces cerevisiae. Eukaryot. Cell 7, 2078–2086 (2008).
Google Scholar
-
Valenzuela, L., Dhillon, N. & Kamakaka, R. T. Transcription independent insulation at TFIIIC-dependent insulators. Genetics 183, 131–148 (2009).
Google Scholar
-
de Boer, C. G. & Taipale, J. Hold out the genome: a roadmap to solving the cis-regulatory code. Nature 625, 41–50 (2024).
Google Scholar
-
Li, Y. et al. Genome-wide Cas9-mediated screening of essential non-coding regulatory elements via libraries of paired single-guide RNAs. Nat. Biomed. Eng. 8, 890–908 (2024).
Google Scholar
-
Castanera, R. et al. Transposable elements versus the fungal genome: impact on whole-genome architecture and transcriptional profiles. PLoS Genet 12, e1006108 (2016).
Google Scholar
-
McDonald, M. C. et al. Transposon-mediated horizontal transfer of the host-specific virulence protein ToxA between three fungal wheat pathogens. MBio 10, e01515–e01519 (2019).
Google Scholar
-
Muszewska, A., Steczkiewicz, K., Stepniewska-Dziubinska, M. & Ginalski, K. Transposable elements contribute to fungal genes and impact fungal lifestyle. Sci. Rep. 9, 4307 (2019).
Google Scholar
-
Yoshida, K. et al. Host specialization of the blast fungus Magnaporthe oryzae is associated with dynamic gain and loss of genes linked to transposable elements. BMC Genomics 17, 370 (2016).
Google Scholar
-
Krishnan, P. et al. Transposable element insertions shape gene regulation and melanin production in a fungal pathogen of wheat. BMC Biol. 16, 78 (2018).
Google Scholar
-
Omrane, S. et al. Plasticity of the MFS1 promoter leads to multidrug resistance in the wheat pathogen Zymoseptoria tritici. mSphere 2, e00393–17 (2017).
Google Scholar
-
Gusa, A. et al. Transposon mobilization in the human fungal pathogen Cryptococcus is mutagenic during infection and promotes drug resistance in vitro. Proc. Natl. Acad. Sci. USA 117, 9973–9980 (2020).
Google Scholar
-
Priest, S. J. et al. Uncontrolled transposition following RNAi loss causes hypermutation and antifungal drug resistance in clinical isolates of Cryptococcus neoformans. Nat. Microbiol. 7, 1239–1251 (2022).
Google Scholar
-
Gusa, A. et al. Genome-wide analysis of heat stress-stimulated transposon mobility in the human fungal pathogen Cryptococcus deneoformans. Proc. Natl. Acad. Sci. USA 120, e2209831120 (2023).
Google Scholar
-
Hess, J. et al. Transposable element dynamics among asymbiotic and ectomycorrhizal Amanita fungi. Genome Biol. Evol. 6, 1564–1578 (2014).
Google Scholar
-
Bucknell, A. H. & McDonald, M. C. That’s no moon, it’s a Starship: giant transposons driving fungal horizontal gene transfer. Mol. Microbiol. 120, 555–563 (2023).
Google Scholar
-
Urquhart, A. S., Vogan, A. A., Gardiner, D. M. & Idnurm, A. Starships are active eukaryotic transposable elements mobilized by a new family of tyrosine recombinases. Proc. Natl. Acad. Sci. USA 120, e2214521120 (2023).
Google Scholar
-
Gluck-Thaler, E. et al. Giant Starship elements mobilize accessory genes in fungal genomes. Mol. Biol. Evol. 39, msac109 (2022).
Google Scholar
-
Vogan, A. A. et al. The Enterprise, a massive transposon carrying Spok meiotic drive genes. Genome Res. 31, 789–798 (2021).
Google Scholar
-
Urquhart, A. S., Chong, N. F., Yang, Y. & Idnurm, A. A large transposable element mediates metal resistance in the fungus Paecilomyces variotii. Curr. Biol. 32, 937–950.e5 (2022).
Google Scholar
-
Gourlie, R. et al. The pangenome of the wheat pathogen Pyrenophora tritici-repentis reveals novel transposons associated with necrotrophic effectors ToxA and ToxB. BMC Biol. 20, 239 (2022).
Google Scholar
-
Bucknell, A. et al. Sanctuary: A Starship transposon facilitating the movement of the virulence factor ToxA in fungal wheat pathogens. bioRxiv https://doi.org/10.1101/2024.03.04.583430 (2024).
-
Gluck-Thaler, E. & Vogan, A. A. Systematic identification of cargo-mobilizing genetic elements reveals new dimensions of eukaryotic diversity. Nucleic Acids Res. 52, 5496–5513 (2024).
Google Scholar
-
Vande Zande, P., Zhou, X. & Selmecki, A. The dynamic fungal genome: polyploidy, aneuploidy and copy number variation in response to stress. Annu. Rev. Microbiol. 77, 341–361 (2023).
Google Scholar
-
Todd, R. T., Wikoff, T. D., Forche, A. & Selmecki, A. Genome plasticity in Candida albicans is driven by long repeat sequences. Elife 8, e45954 (2019).
Google Scholar
-
Raffaele, S. & Kamoun, S. Genome evolution in filamentous plant pathogens: why bigger can be better. Nat. Rev. Microbiol. 10, 417–430 (2012).
Google Scholar
-
Bowyer, P., Currin, A., Delneri, D. & Fraczek, M. G. Telomere-to-telomere genome sequence of the model mould pathogen Aspergillus fumigatus. Nat. Commun. 13, 5394 (2022).
Google Scholar
-
Zhang, Z. et al. Complete telomere-to-telomere genomes uncover virulence evolution conferred by chromosome fusion in oomycete plant pathogens. Nat. Commun. 15, 4624 (2024).
Google Scholar
-
van der Burgt, A., Karimi Jashni, M., Bahkali, A. H. & de Wit, P. J. G. M. Pseudogenization in pathogenic fungi with different host plants and lifestyles might reflect their evolutionary past. Mol. Plant Pathol. 15, 133–144 (2014).
Google Scholar
-
Jackson, A. P. et al. Comparative genomics of the fungal pathogens Candida dubliniensis and Candida albicans. Genome Res. 19, 2231–2244 (2009).
Google Scholar
-
Lafontaine, I. & Dujon, B. Origin and fate of pseudogenes in Hemiascomycetes: a comparative analysis. BMC Genomics 11, 260 (2010).
Google Scholar
-
Harrison, P. et al. A small reservoir of disabled ORFs in the yeast genome and its implications for the dynamics of proteome evolution. J. Mol. Biol. 316, 409–419 (2002).
Google Scholar
-
Vitiello, M. & Poliseno, L. CRISPR/Cas technologies applied to pseudogenes. in Pseudogenes: Functions and Protocols (ed. Poliseno, L.) 265–284 (Springer, 2021).
-
Cheng, H. et al. Small open reading frames: current prediction techniques and future prospect. Curr. Protein Pept. Sci. 12, 503–507 (2011).
Google Scholar
-
Mundodi, V., Choudhary, S., Smith, A. D. & Kadosh, D. Global translational landscape of the Candida albicans morphological transition. G3 11, jkaa043 (2021).
Google Scholar
-
Li, Q.-R. et al. Revisiting the Saccharomyces cerevisiae predicted ORFeome. Genome Res. 18, 1294–1303 (2008).
Google Scholar
-
Saeki, N. et al. N-terminal deletion of Swi3 created by the deletion of a dubious ORF YJL175W mitigates protein burden effect in S. cerevisiae. Sci. Rep. 10, 9500 (2020).
Google Scholar
-
Sahu, P. K., Salim, S., Pp, M., Chauhan, S. & Tomar, R. S. Reverse genetic analysis of yeast YPR099C/MRPL51 reveals a critical role of both overlapping ORFs in respiratory growth and MRPL51 in mitochondrial DNA maintenance. FEMS Yeast Res. 19, foz056 (2019).
Google Scholar
-
Rech, G. E., Sanz-Martín, J. M., Anisimova, M., Sukno, S. A. & Thon, M. R. Natural selection on coding and noncoding DNA sequences is associated with virulence genes in a plant pathogenic fungus. Genome Biol. Evol. 6, 2368–2379 (2014).
Google Scholar
-
Ellwood, S. R., Syme, R. A., Moffat, C. S. & Oliver, R. P. Evolution of three Pyrenophora cereal pathogens: recent divergence, speciation and evolution of non-coding DNA. Fungal Genet. Biol. 49, 825–829 (2012).
Google Scholar
-
Maunakea, A. K. et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466, 253–257 (2010).
Google Scholar
-
Maroc, L., Shaker, H. & Shapiro, R. S. Functional genetic characterization of stress tolerance and biofilm formation in Nakaseomyces (Candida) glabrata via a novel CRISPR activation system. mSphere 9, e0076123 (2024).
Google Scholar
-
Gervais, N. C. et al. Development and applications of a CRISPR activation system for facile genetic overexpression in Candida albicans. G3 13, jkac301 (2023).
Google Scholar
-
Ciurkot, K., Gorochowski, T. E., Roubos, J. A. & Verwaal, R. Efficient multiplexed gene regulation in Saccharomyces cerevisiae using dCas12a. Nucleic Acids Res. 49, 7775–7790 (2021).
Google Scholar
-
Després, P. C., Dubé, A. K., Seki, M., Yachie, N. & Landry, C. R. Perturbing proteomes at single residue resolution using base editing. Nat. Commun. 11, 1871 (2020).
Google Scholar
-
Jing, X. et al. Implementation of the CRISPR-Cas13a system in fission yeast and its repurposing for precise RNA editing. Nucleic Acids Res. 46, e90 (2018).
Google Scholar
Acknowledgements
N.C.G. is supported by an Ontario Graduate Scholarship and R.S.S. is supported by a Tier II Canada Research Chair from the Natural Sciences and Engineering Research Council of Canada (NSERC). The authors would like to acknowledge other important research that was not cited due to space limitations.
Author information
Authors and Affiliations
Contributions
N.C.G. conceptualized this work and made the figures. N.C.G. and R.S.S. wrote and revised the manuscript together.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Neeraj Chauhan, Asiya Gusa and the other, anonymous, reviewer for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
Reprints and permissions
About this article
Cite this article
Gervais, N.C., Shapiro, R.S. Discovering the hidden function in fungal genomes.
Nat Commun 15, 8219 (2024). https://doi.org/10.1038/s41467-024-52568-z
-
Received: 25 April 2024
-
Accepted: 11 September 2024
-
Published: 19 September 2024
-
DOI: https://doi.org/10.1038/s41467-024-52568-z
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.