Abstract
The chemical classes of semicarbazones, thiosemicarbazones, and hydrazones are present in various compounds, each demonstrating diverse biological activities. Extensive studies have revealed their potential as schistosomicidal agents. Thiosemicarbazones, in particular, have shown inhibitory effects on Schistosoma mansoni’s cathepsin B1 enzyme (SmCB1), which plays a crucial role in hemoglobin degradation within the worm’s gut and its nutrition processes. Consequently, SmCB1 has emerged as a promising target for novel schistosomiasis therapies. Moreover, chloroquinoline exhibits characteristics in its aromatic structure that hold promise for developing SmCB1 inhibitors, along with its interaction with hemoglobin’s heme group, potentially synergizing against the parasite’s gut. In this context, we report the synthesis of 22 hybrid analogs combining hydrazones and quinolines, evaluated against S. mansoni. Five of these hybrids demonstrated schistosomicidal activity in vitro, with GPQF-8Q10 being the most effective, causing worm mortality within 24 h at a concentration of 25 µM. GPQF-8Q8 proved to be the most promising in vivo, significantly reducing egg presence in feces (by 52.8%) and immature eggs in intestines (by 45.8%). These compounds exhibited low cytotoxicity in Vero cells and an in in vivo animal model (Caenorhabditis elegans), indicating a favorable selectivity index. This suggests their potential for the development of new schistosomiasis therapies. Further studies are needed to uncover specific target mechanisms, but these findings offer a promising starting point.
Introduction
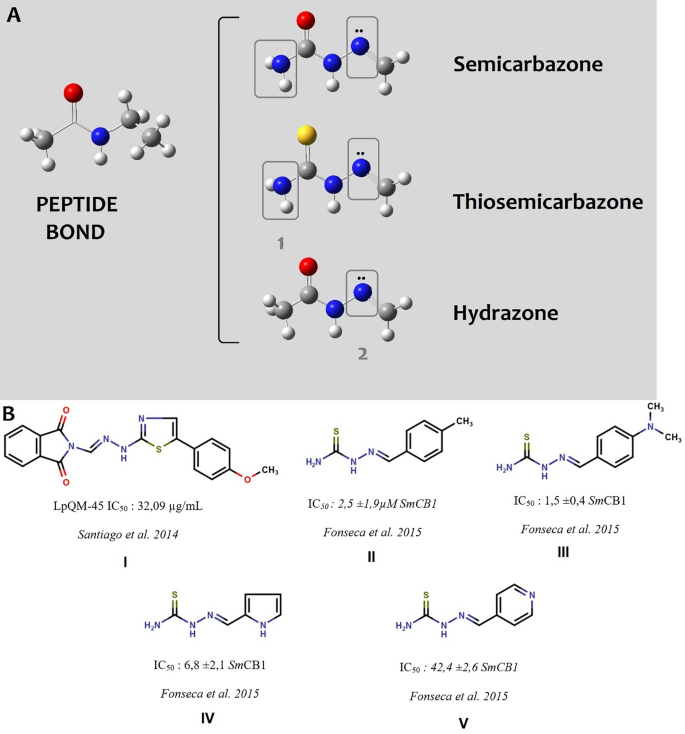
Semicarbazones, thiosemicarbazones, and hydrazones are chemical classes of compounds with a broad range of biological activities. Due to their resemblance to the peptide bonds in proteins, they are regarded as privileged chemical groups, since they mislead macromolecules that initially identify peptide connections. Moreover, they present additional points for intermolecular interaction, which allows them to have greater affinity for the target (Fig. 1A).

Semicarbazone, thiosemicarbazone, and hydrazone scaffolds and their active representatives against Schistosoma mansoni cathepsin B1. (A) Semicarbazones, thiosemicarbazones, and hydrazones similarities with peptide bonds and the extra points for potential new interactions highlighted in rectangles (1: hydrogen bond donor/acceptor and 2: hydrogen bond acceptor) with macromolecular targets. Atoms are sketched as ball-sticks and colored following the pattern: blue: nitrogen; red: oxygen; gray: carbon, yellow: sulfur and white: hydrogen. (B) SmCB1 inhibitors from the chemical classes of the semicarbazone and thiosemicarbazones described in the literature.
In 2014, Santiago et al.1 conducted in vitro studies with ten thiosemicarbazones and hydrazones. They selected an analog, the LpQM-45, which significantly reduced worm motility, male–female pairing, and oviposition after 144 h, with an IC50 value of 32.09 μg/mL. However, the macromolecular target of these compounds, was not determined in this study. In 2015, Fonseca et al.2 synthesized and tested 29 thiosemicarbazones with a direct action on cathepsin B1 from Schistosoma mansoni (SmCB1), revealing a set of five compounds with IC50 values under 10 μM. However, all the other analogs, showed only moderate inhibitory activity (Fig. 1B). Cathepsin B1 from S. mansoni is a peptidase responsible for hemoglobin degradation in the worm’s gut and, is the main protease involved in nutrition. This protease is crucial not only for worm survival but also, in the case of females, for oviposition, which ensures the continuity of the biological cycle3.
As SmCB1 enzyme is considered a validated target, it warrants further efforts in the search for new inhibitors. Upon examining the structures presented by Fonseca et al. structures, it becomes evident that the majority of them also feature nitrogen in either the ortho or para position of the aromatic ring, specifically in relation to the azomethine group within their structures. While it appears that the activity of these compounds is not solely dependent on the presence of this nitrogen atom, it is noteworthy that most of the highly active compounds do indeed contain it. Taken this observation into account an in pursuit of rational molecular modifications to these structures, we proposed incorporating 4-amino-7-chloroquinoline as the aromatic system attached to hydrazones analogs of the compounds studied by Fonseca et al.2.
The rationale behind this proposal is based on the knowledge that chloroquine, a well-known widely used drug against the protozoa Plasmodium spp., features an aromatic nitrogen in the same position. It is also well-established that chloroquine interacts with the heme group from hemoglobin4. Given that the primary source of nutrients for S. mansoni is the hemoglobin, and SmCB1 is responsible for its degradation, hybrid compounds combining the characteristics of both compound classes could potentially achieve a synergistic effect within the parasite’s digestive system.
Here, we present the results of synthesizing these compounds and conducting in vitro and in vivo biological screening against adult male and female S. mansoni worms.
Results and discussions
A series of 22 analogs of 7‐chloro‐4‐[2‐(phenylmethylidene)hydrazin‐1‐yl]quinoline were synthesized through the coupling of 7‐chloro‐4‐hydrazinylquinoline with various aldehydes or ketones, as described in Methods Session and illustrated in Fig. 2. In general, the compounds were obtained in solid form with good purity, which was confirmed through 1H-NMR and melting point analysis, and in moderate to good yields. As expected, Aldehyde derivatives, were obtained in higher yields than the ketone derivatives.

Synthetic approach to obtain the 22 analogs of 7‐chloro‐4‐[2‐(phenylmethylidene)hydrazin‐1‐yl]quinoline. General synthesis of 7-chloro-4-hydrazinylquinoline (1) and 7-chloro-4-hydrazinylquinoline derivatives (2). Reagents and conditions: (a) ethanol, reflux 80 °C, for 15 h; (b) corresponding aldehydes and ketones, ethanol/HOAc 25% or MeOH/HOAc 25%, reflux 80 °C, 3-24 h. In specific cases (GPQF-8Q1, 8Q22, 8Q23, and 8Q25) the reaction occurred at room temperature for 24 h.
Table 1 lists the structures of the compounds in the series, along with their yields and the results of the initial in vitro trial conducted at 50 micromolars.
The ease of synthesis, along with good yields, the use of relatively inexpensive reagents, and the production of solid compounds, all represent desirable characteristics for drug candidates targeting neglected diseases like schistosomiasis.
All compounds were subjected to an initial trial at 50 µM. In the context of testing compounds against S. mansoni, only chemical compounds or natural extracts that demonstrated any activity at this concentration were considered significant for further in vitro and in vivo testing. Accordingly, five out of the 22 final compounds met these criteria and were selected to follow with the experimental investigations.
This first trial also uncovered some intriguing insights into the relationships between the chemical properties of the compounds and their biological behavior. Notably, all ketone derivatives proved to be inactive against the adult worms, at least within the established concentrations limits of the test. Even compounds with similar overall structures but derived from equivalent aldehydes or ketones, such as the pairs GPQF-8Q9/GPQF-8Q17, and GPQF-8Q10/GPQF-8Q18, exhibited contrasting biological responses at 50 µM, with only the aldehydes derivatives displaying activity. This strongly suggests the presence of a macromolecular target with specific stereochemical and/or volume requirements. The ketone derivatives, which possess a three-dimensional structure distinct from the typically flat structure of the azomethines derived from aldehydes, were inactive. These initial results point towards a stereo-specific binding site, where only the flat derivatives could exert significant inhibitory activity.
The five active analogs were subsequently subjected to in vitro assays at lower concentrations. The results, which include reductions in worm motor activity categorized as normal, slight, significant, or absent and are separated by sex, are presented in Fig. 3.

The viability of adult S. mansoni following incubation with five active analogs.
All compounds exhibited significant interference with worm motility at concentrations of 50 µM or lower. Distinctions in their biological behavior became apparent at 25 µM, where some compounds demonstrated effective interference with motility and lethality against the worms (Fig. 4).

In vitro antischistosomal activity results after exposure to the compounds at 25 µM. Control = DMSO.
With regard to lethality, GPQF-8Q10 proved to be the most effective analog in vitro, killing the worms within the first 24 h at 25 µM. A noticeable reduction in motility followed a similar pattern. It appears that there may be differences in sensitivity between males and females, although these results do not provide a detailed understanding of how each gender responds to the treatment. Notably, females appeared to be affected by all the active analogs to varying degrees and at different time intervals, with the exception of GPQF-8Q2.
Another noteworthy observation is the compounds require some time to exert their lethal effects, as none of them killed the worms immediately. This time-dependent activity aligns with the design concept of these compounds, as their potential inhibition of SmCB1 suggests that they induced death by interfering with nutrition, a process that naturally takes some time to occur.
The active compounds were subjected to testing against Vero cells (African green monkey kidney cells), a commonly employed cell line in toxicity studies, especially in the realm of antischistosomal drug discovery13,17. The cytotoxicity concentration that led to a 50% cell death rate (CC50) was determined. The results, including the effective concentration that caused 50% inhibition (EC50) and the selectivity index, are summarized in Table 2.
The measured EC50 values confirm the initial in vitro observations. The Vero cells assay indicated low toxicity of these compounds against these cells and the calculated selectivity indexes suggests that good selectivity could be achieved.
The next step in this investigation was the in vivo tests (Fig. 5).

Biological in vivo results after 400 mg/kg single dose of the compounds. Efficacy of analogs of the 7‐chloro‐4‐[2‐(phenylmethylidene)hydrazin‐1‐yl]quinoline in a schistosomiasis animal model. Drugs were administered orally using a single dose of 400 mg/kg to S. mansoni-infected mice. On day 56 post-infection, all animals were humanely euthanized. Parasite burdens were determined by sex (male and female parasites). Egg burdens were determined by counting eggs in the feces (Kato-Katz technique) and in the tissue (oogram analysis in the intestine). Data are presented as the mean ± SD (n = 5 per group). The numbers represent the percentages of worms or egg reduction vs. infected untreated control. The partition coefficient (CLogP) is indicated just below the chemical structures. *P < 0.05; **P < 0.01 compared to the control group infected and treated with the vehicle.
Remarkably, the best-performing compound in in vitro tests, GPQF-8Q10, did not exhibit the same level of effectiveness in vivo. It might be tempting to associate this lack of in vivo activity with its high partition coefficient (CLogP 5.06). However, this does not appear to be the case, as the analog GPQF-8Q2, which had the highest CLogP value (5.17), showed the best reduction in oviposition. The most significant reductions in worm burden were achieved by compounds GPQF-8Q8 and GPQF-8Q9, with reductions of 50.8 and 58.7%, respectively. GPQF-8Q8 displayed the most comprehensive in vivo biological activity, as it was also the most effective compound in interfering with egg production. It led to reductions in both eggs found in feces (52.8%) and immature eggs found in the intestines (45.8%). The presence of the thiophene ring in this compound also contributed positively to lipophilicity, which could suggest that this factor plays a role in the action on oviposition.
Even the more “hydrophilic” compounds, GPQF-8Q9 and GPQF-8Q11 (both having a CLogP of 4.26 as they are position isomers), exhibited intriguing in vivo biological behaviors. Interestingly, only GPQF-8Q9 showed significant in vivo activity, suggesting the presence of an important steric factor related to theses structures. Ortho substituents are well-known to influence the conformation of rings, forcing them out of the plane of the molecule, in this case, the plane of the 7-chloroquinoline ring. This could explain why GPQF-8Q11 did not exhibit any in vivo activity at all. Since the only difference between these two compounds is the steric positioning of hydroxy group, the significantly different biological responses to both compounds strongly indicate the presence of an endogenous receptor with selective steric specificities.
Another noteworthy observation is that the two best-performing in vivo, GPQF-8Q8 and GPQF-8Q9, were also the only two compounds that predominantly exhibited in vitro activity against females. In a study by Liu et al.5 in 2014, it was demonstrated that S. japonicum expresses a network of proteases in its gut, with Cathepsin B1 being the predominant enzyme. Importantly, they observed that this enzyme was upregulated in female gut. Therefore, the preference of GPQF-8Q8 and GPQF-8Q9 for acting on females could be a strong indication that SmCB1 is indeed the target of these compounds.
It is important to highlight, however, that the SmCB1 could not be the only possible macromolecular target since the in vitro tests do not guarantee the specificity for only one target. Only enzymatic inhibitory assays would prove that this enzyme would be a real target for these compounds, and they have already been performed by our group. But even in this case the selectivity for just one target could not be assured.
Caenorhabditis elegans is a nematode commonly employed as an in vivo toxicity model 6,7. Its utility extends to research aiming to discover new anthelmintic agents 8,9. In order to delve deeper into the potential toxicity of analogs of 7‐chloro‐4‐[2‐(phenylmethylidene)hydrazin‐1‐yl]quinoline, we assessed the survival of C. elegans exposed to various compound concentrations. As shown in Table 3, none of the compounds exhibited any toxic effects, and all nematodes displayed a sinusoidal shape and mobility comparable to the untreated animals.
Conclusion
Considering the results reported herein, the 7‐chloro‐4‐[2‐(phenylmethylidene)hydrazin‐1‐yl]quinoline scaffold appears to be a promising starting point for the development of new antischistosomal drugs. Among the 22 tested compounds, five demonstrated schistosomicidal activity in the in vitro assay, with GPQF-8Q10 emerging as the most potent compound at 25 µM, leading to the death of all worms within 72 h. Notably, this compound displayed values of 13.3 and 15.6 µM against male and female worms, respectively.
In the in vivo assay, GPQF-8Q8 displayed the most comprehensive biological activity, reducing both the number of eggs found in feces (52.8%) and immature eggs found in the intestines (45.8%). The compounds also demonstrated low cytotoxicity against Vero cells and a favorable selectivity index. Furthermore, none of the compounds exhibited any toxic effect in the in vivo toxicity assay using the C. elegans model.
Although further studies are needed to determine the target, the collective findings strongly suggest the potential of the hydrazone quinoline scaffold in the quest for new antischistosomal leads.
Methods
Chemistry
Reagents and solvents were obtained from commercial suppliers without the need for further purification. Chemical reactions were monitored by thin layer chromatography (TLC) using Merck 60 F254 Silica gel on TLC aluminum foils, hexane/ethyl acetated 1:2 was employed as eluent and the plates were visualized by ultraviolet (UV) irradiation (254 nm). Melting point analyses were conducted in triplicate using a Marte científica PDF III apparatus. Infrared (IR) analyses were carried out using an IR Affinity-1 Shimadzu Fourier Transform Infrared Spectrophotometer. Samples were prepared with anhydrous KBr, and the analyses covered the wave number region between 4000 and 400 cm−1. 1H-NMR analyses were performed on a Bruker-300 Ultrashield operating at 300 MHz, with solvents DMSO-d6 or CDCl3. The chemical shift values (δ) were expressed in parts per million (ppm), employing tetramethylsilane (TMS) as the internal reference. The following abbreviations were used to describe the multiplicities: s = singlet; d = doublet; dd = double doublet; t = triplet; m = multiplet. Coupling constants (J) were described in Hertz.
All the structural data collected are provided in the Supplementary Information (Fig. S1–S46).
The synthesis of the intermediate 7-chloro-4-hydrazinylquinoline was conducted following a previously described methodology in the literature10,11,12. A total of 5 mmol of 4,7-Dichloroquinoline was dissolved in 10 mL of ethanol and placed in a round-bottom flask equipped with a reflux system. After 5 min of stirring, 25 mmol of hydrazine hydrate 80% (w/v) was added to the reaction. The solution was then heated under reflux for 15 h at 80–90 °C. After this period, the solution was refrigerated for approximately 8 h. The resulting dark yellow precipitate was washed with cold distilled water and filtered under reduced pressure. The crude compound was subsequently recrystallized from ethanol and a yellow crystalline solid was obtained.
7-Chloro-4-hydrazinylquinoline
Prepared in accordance with the synthetic methodology previously described. Yellow crystalline solid. Yield 80%. M.P. 220–224 °C (lit.11 225–226°C). 1H-NMR-(DMSO-d6)—δ = ppm: 4.45 (sl, 2H, H12); 6.86 (d, 1H, J = 6 Hz, H3); 7.38 (dd, 1H, J1 = 9Hz, J2 = 3 Hz, H7); 7.75 (s, 1H, H9); 8.16 (d, 1H, J = 9 Hz, H6); 8.40 (d, 1H, J = 9 Hz, H2) 8.59 (sl, 1H, H11);. IR—νNH2 and νNH 3282 and 3253 cm-1; νCH 3053 cm−1; νC = C 1612 cm−1; νC = N 1570 cm−1.
General procedure: quinoline derivatives
The quinoline derivatives were obtained following the synthetic methodology reported in the literature12,13,14. First, (1 mmol) of corresponding aldehyde or ketone was introduced into a round-bottom flask, and in most cases*, approximately 8 mL of a ethanol/acetic acid 25% solution was added to the round-bottom flask. The solution was kept under agitation until the complete solubilization of the corresponding reagents. Then, (1 mmol) of 7-chloro-4-hydrazinylquinoline was added, and the reaction mixture was heated under reflux** (about 80 °C) for about 3–17 h. After the reflux complete, the resulting mixture was concentrated under reduced pressure, and the crude compounds were washed with cold water and cold ethyl ether, then filtered under reduced pressure to obtain the desired products.
*In some cases, the solvent employed was ethanol (GPQF-8Q1) or a solution of MeOH/HAOc 25% (GPQF-8Q6).
**In the case of GPQF-8Q1, 8Q22, 8Q24, and 8Q25, the reaction was kept at room temperature for 24 h.
4-[(2E)-2-benzylidenehydrazinyl]-7-chloroquinoline (GPQF-8Q1)
Prepared following the general synthetic procedure of quinoline derivatives, which involved using 7-chloro-4-hydrazinylquinoline (1mmol) and benzaldehyde (1 mmol) as reactants, ethanol was employed as the solvent, and the reaction was maintained at room temperature for 24 h. Yellow solid. Yield 52%. M.P. 220–224 °C (lit.13 223–225°C). 1H-NMR-(DMSO-d6)—δ = ppm: 7.37 (d, 1H, J = 6 Hz, H3); 7.42–7.51 (m, 3H, H16, H17 H18); 7.57 (dd, 1H, J1 = 9Hz, J2 = 3, H7); 7.81 (d, 2H, J = 6 Hz; H15 e H19); 7.86 (s, 1H, H9); 8.42 (d, 1H, J = 9 Hz, H6) 8.44 (s, 1H, H13); 8.47 (d, 1H, J = 6 Hz, H2) . IR—νNH 3446 cm−1; νCH 3188 and 2891 cm−1; νC = C 1618 cm−1; νC = N 1577 cm−1.
7-Chloro-4-[(2E)-2-(4-chlorobenzylidene)hydrazinyl]quinoline (GPQF-8Q2)
Prepared in accordance with the general synthetic procedure for quinoline derivatives, which involved using 7-chloro-4-hydrazinylquinoline (1mmol) and 4-chlorobenzaldehyde (1 mmol), a solution of ethanol and acetic acid 25% was employed as the solvent. The reaction was conducted under reflux at 80 °C during 3 h. Orange solid. Yield 75%. M.P. 320–324 °C. 1H-NMR-(DMSO-d6)—δ = ppm: 7.60 (d, 2H, J = 9, H16, H18), 7.65 (d, 1H, J = 6, H3), 7.88 (d, 1H, J = 9 Hz, H7), 7.94 (d, 2H, J = 9 Hz, H15, H19), 8.07 (d, 1H, J = , H9), 8.69 (d, 1H, J = 6 Hz, H6), 8.75–8.78 (m, 2H, H13, H2). IR—νNH 3446 cm−1; νCH 2875 and 2698 cm−1; νC = C 1627 cm−1; νC = N 1612 cm−1.
7-Chloro-4-[(2E)-2-(4-nitrobenzylidene)hydrazinyl]quinoline (GPQF-8Q3)
7-Chloro-4-hydrazinylquinoline (1mmol) and 4-nitrobenzaldehyde (1 mmol) were coupled in the presence of ethanol and acetic acid 25% solution. The reaction mixture was refluxed at 80 °C for 18 h, following the general synthetic procedure. Pale-yellow solid. Yield 59%. M.P. 348–350 °C. 1H-NMR-(DMSO-d6)—δ = ppm: 7.74 (d, 1H, J = 6Hz, H3), 7.93 (dd, 1H, J1 = 9Hz, J2 = 3Hz, H7), 8.08 (s, 1H, H9), 8.19 (d, 2H, J = 9Hz, H15, H19), 8.36 (d, 2H, J = 9Hz, H16, H18), 8.73–8.81 (m, 2H, H2, H6, H13). IR—νNH 3446 cm−1; νCH 2646 cm−1; νC = C 1614 cm−1.
4-{(E)-[2-(7-chloroquinolin-4-yl)hydrazinylidene]methyl}-2-methoxyphenol (GPQF-8Q4)
7-Chloro-4-hydrazinylquinoline (1 mmol) and 4-hydroxy-3-methoxybenzaldehyde (1 mmol) were coupled following the general synthetic procedure, in the presence of ethanol/acetic acid 25% solution and the reaction was maintained under reflux at 80 °C for 3 h. Yellow solid. Yield 63%. M.P. 284–288 °C (lit.10 272–275 °C). 1H-NMR-(DMSO-d6)—δ = ppm: 3.89 (s, 3H, H22); 6.94 (d, 1H, J = 6 Hz, H3); 7.25 (d, 1H, J = 3Hz, H15); 7.45 (s, 1H, H19); 7.61 (d, 1H, J = 9 Hz, H16); 7.84 (d, 1H, J = 9 Hz, H7); 8.10 (s, 1H, H9); 8.61 (d, 1H, J = 6 Hz, H6); 8.77 (s, 1H, H13); 8.91 (d, 1H, J = 9 Hz, H2); 9.96 (sl, 1H, H11); 12.98 (sl, 1H, H23). IR—νOH and νNH 3421 cm−1; νCH 3209 cm−1; νC = C 1608 cm−1; νC = N 1589 cm−1.
7-Chloro-4-{(2E)-2-[(5-nitrofuran-2-yl)methylidene]hydrazinyl}quinoline (GPQF-8Q6)
Prepared following the general synthetic procedure for quinoline derivatives, which involved using 7-chloro-4-hydrazinylquinoline (1mmol) and 5-nitrofuran-2-carbaldehyde (1 mmol), a solution of methanol and acetic acid 25% was used as the solvent. The reaction was maintained under reflux at 80 °C for 17 h. Mustard yellowish solid. Yield 58%. M.P. 284–286 °C (lit.15 238–240 °C). 1H-NMR-(DMSO-d6)—δ = ppm: 7.46 (d, 1H, J = 3Hz, H4), 7.52 (d, 1H, J = 6Hz, H12), 7.83 (d, 1H, J = 3 Hz, H4), 7.86 (d, 1H, J = 6Hz, H18), 8.07 (s, 1H, H16), 8.71 (d, 1H, J = 6 Hz, H19), 8.76 (s, 1H, H8) 8.82 (d, 1H, J = 9Hz, H13). IR—νNH 3446 cm−1; νCH 3082 and 2646 cm−1; νC = C 1614 cm−1; νC = N 1589 cm−1.
7-Chloro-4-[(2E)-2-(thiophen-2-ylmethylidene)hydrazinyl]quinoline (GPQF-8Q8)
7-Chloro-4-hydrazinylquinoline (1mmol) and thiophene-2-carbaldehyde (1 mmol) were coupled following the general synthetic procedure, in the presence of a solution of ethanol and acetic acid 25%. The reaction mixture was maintained under reflux 80 °C for 16 h. Yellowish bright solid. Yield 61%. M.P. 224–226 °C (lit.15 231–232 °C). 1H-NMR-(DMSO-d6)—δ = ppm: 7.15–7.19 (m, 2H, H3, H10), 7.49 (d, 1H, J = 3Hz, H4), 7,57 (dd, 1H, J1 = 9Hz, J2 = 3Hz, H16), 7.66 (d, 1H, J = 6 Hz, H2), 7.82 (s, 1H, H14), 8.37–8.43 (m, 2H, H11, H17), 8.66 (s, 1H, H6). IR—νNH 3444 cm−1; νCH 3074cm−1; νC = C 1610 cm−1; νC = N 1550 cm−1.
4-{(E)-[2-(7-chloroquinolin-4-yl)hydrazinylidene]methyl}phenol (GPQF-8Q9)
7-Chloro-4-hydrazinylquinoline (1mmol) and 4-hydroxybenzaldehyde (1 mmol) were coupled following the general synthetic procedure, in the presence of a solution of ethanol and acetic acid 25% and maintained under reflux at 80 °C for 16 h. Orange solid. Yield 77%. M.P. 280–284 °C (lit.10 233–235 °C). 1H-NMR-(DMSO-d6)—δ = ppm: 6.85 (d, 2H, J = 9Hz, H16, H18), 7.26 (d, 1H, J = 3 Hz, H3), 7.51 (d, 1H, J = 6 Hz, H7), 7.62 (d, 2H, J = 9 Hz, H15, H19), 7.82 (sl, 1H, H9), 8.31 (s, 1H, H13), 8.35 (d, 1H, J = 12Hz, H6); 8.47 (d, 1H, J = 12 Hz, H2) 9.97 (sl, 1H, H21), 11,03 (sl, 1H, H11). ). IR—νO-H and νNH 3425 cm−1; νCH 3030cm−1; νC = C 1608 cm−1; νC = N 1552 cm−1.
7-Chloro-4-[(2E)-2-(4-methylbenzylidene)hydrazinyl]quinoline (GPQF-8Q10)
Prepared following the general synthetic procedure, which involved using 7-chloro-4-hydrazinylquinoline (1mmol) and 4-methylbenzaldehyde (1 mmol), in the presence of a solution of ethanol and acetic acid 25%. The reaction was maintained under reflux at 80 °C for 7 h. Sharp range crystals. Yield 96%. M.P. 190–192 °C (lit.12 240–244 °C). 1H-NMR-(DMSO-d6)—δ = ppm: 2.36 (s, 3H, H21), 7.29 (d, 2H, J = 9Hz, H16, H18), 7.34 (d, 1H, J = 6Hz, H3), 7.56 (dd, 1H, J1 = 9Hz, J2 = 3Hz, H7), 7.69 (d, 2H, J = 9Hz, H15, H19), 7.84 (d, 1H, J = 3Hz, H9), 8.38–8.41 (m, 2H, H6, H13), 8.45 (d, 1H, J = 6Hz, H2). IR—νNH 3446 cm−1; νCH 3022cm−1; νC = C 1618 cm−1; νC = N 1579 cm−1.
2-{(E)-[2-(7-chloroquinolin-4-yl)hydrazinylidene]methyl}phenol (GPQF-8Q11)
Prepared in accordance with general synthetic procedure. 7-chloro-4-hydrazinylquinoline (1mmol) and 2-hydroxylbenzaldehyde (1 mmol) were coupled using a solution of ethanol and acetic acid 25%. The reaction was conducted under reflux at 80 °C for 17 h. Golden yellow solid. Yield 96%. M.P. 222–226 °C (lit.13 150–151 °C). 1H-NMR-(DMSO-d6)—δ = ppm: 6.90–6.95 (m, 3H, H3, H16, H18), 7.27 (t, 1H, J = 7.5Hz, H17), 7.48 (d, 1H, J = 9Hz, H7), 7.69–7.70 (m, 2H, H6, H19), 8.17 (s, 1H, H9), 8.38 (d, 1H, J = 9Hz, H2), 8.73 (s, 1H, H13). IR—νO-H and νNH 3427 cm−1; νCH 2553cm−1; νC = C 1622 cm−1; νC = N 1554 cm−1.
7-Chloro-4-[(2E)-2-(4-fluorobenzylidene)hydrazinyl]quinoline (GPQF-8Q12)
Prepared in accordance with general synthetic procedure. 7-chloro-4-hydrazinylquinoline (1mmol) and 4-fluorobenzaldehyde (1 mmol) were coupled, using a solution of ethanol and acetic acid 25%. The reaction was conducted under reflux at 80 °C for 17 h. Mustard yellow solid. Yield 86%. M.P. 236–241 °C (lit.13 225–226 °C). 1H-NMR-(DMSO-d6)—δ = ppm: 7.28–7.35 (m, 3H, H3, H15, H19), 7.56 (dd, 1H, J1 = 9Hz, J2 = 3Hz, H7), 7.84–7.89 (m, 3H, H9, H16, H18), 8.37–8.42 (m, 3H, H2, H6, H13). IR—νNH 3421 cm−1; νCH 3209cm−1; νC = C 1608 cm−1; νC = N 1571 cm−1.
7-Chloro-4-[(2E)-2-(3,4-dichlorobenzylidene)hydrazinyl]quinoline (GPQF-8Q13)
7-Chloro-4-hydrazinylquinoline (1mmol) and 3,4-dichlorobenzaldehyde (1 mmol) were coupled, using a solution of ethanol and acetic acid 25% employed as the solvent, and kept under reflux at 80 °C during 16 h, following the general synthetic procedure. Light yellow solid. Yield 43%. M.P. 320 °C (decomposition). 1H-NMR-(DMSO-d6)—δ = ppm: 7.70 (d, 1H, J = 9 Hz, H3); 7.79 (d, 1H, J = 6Hz, H16) 7.84–7.92 (m, 2H, H7, H15), 8.03 (s, 1H, H19) 8.19 (s, 1H, H9) 8.63–8.69 (m, 3H, H2, H6, H13). IR—νNH 3414 cm−1; νCH 2879cm−1; νC = C 1616 cm−1; νC = N 1589 cm−1.
4-{(E)-[2-(7-chloroquinolin-4-yl)hydrazinylidene]methyl}benzene-1,3-diol (GPQF-8Q14)
7-chloro-4-hydrazinylquinoline (1mmol) and 3,4-dihydroxybenzaldehyde (1 mmol) were coupled using a solution of ethanol and acetic acid 25% as the solvent. The reaction was conducted under reflux at 80 °C for 16 h, following the general synthetic procedure. Yellow solid. Yield 55%. M.P. 340 °C (decomposition). 1H-NMR-(DMSO-d6)—δ 6.40 (d, 1H, J = 9Hz, H3), 6.45 (s, 1H, H16), 7.46 (d, 1H, J = 9 Hz, H7), 7.75 (d, 1H, J = 9Hz, H18), 7.82 (d, 1H, J = 9Hz, H19), 8.08 (s, 1H, H9), 8,57 (d, 1H, J = 6Hz, H6), 8.81 (d, 1H, J = 9Hz, H2) 9.01 (s, 1H, H13), 10.13 (s, 1H, H22), 10.36 (s, 1H, H21). IR—νO-H and νNH 3427 cm−1; νCH 2897 cm−1; νC = C 1631 cm−1; νC = N 1606 cm−1.
7-Chloro-4-[(2E)-2-(1-phenylethylidene)hydrazinyl]quinoline (GPQF-8Q16)
Prepared following the general synthetic procedure, which involved using 7-chloro-4-hydrazinylquinoline (1mmol) and 1-phenylethanone (1 mmol) as reactants, and a solution of ethanol and acetic acid 25% as the solvent. The reaction was maintained under reflux at 80 °C for 16 h. Light yellow solid. Yield 52%. M.P. 302–306 °C. 1H-NMR-(DMSO-d6)—δ = ppm: 2.65 (s, 3H, H14), 7.49–7.55 (m, 4H, H3, H18, H19, H20), 7.84 (dd, 1H, J1 = 9Hz, J2 = 3Hz, H7), 8.00 (m, 2H, H17, H21), 8.18 (s, 1H, H9), 8.64 (d, 1H, J = 6Hz, H6), 8.82 (d, 1H, J = 9Hz, H2). IR—νNH 3408 cm−1; νCH 2767cm−1; νC = C 1633 cm−1; νC = N 1606 cm−1.
4-{(1E)-1-[2-(7-chloroquinolin-4-yl)hydrazinylidene]ethyl}phenol (GPQF-8Q17)
7-Chloro-4-hydrazinylquinoline (1mmol) and 1-(4-hydroxyphenyl)ethanone (1 mmol) were prepared following the general synthetic procedure, using a solution of ethanol and acetic acid 25% as the solvent. The reaction was conducted under reflux at 80 °C for 16 h. Yellow solid. Yield 54%. M.P. 320 °C (decomposition). 1H-NMR-(DMSO-d6)—δ = ppm: 2.57 (s, 3H, H14), 6.90 (d, 2H, J = 9Hz, H17, H21), 7.45 (d, 1H, J = 9Hz, H3), 7.81 (dd, 1H, J1 = 9Hz, J2 = 3Hz, H7), 7.87 (d, 2H, J = 9Hz, H18, H20), 8.13 (s, 1H, H9), 8.60 (d, 1H, J = 9Hz, H6), 8.87 (d, 1H, J = 9Hz, H2), 10.12 (s, 1H, H22). IR—νO-H and νNH 3209 cm−1; νCH 2787cm−1; νC = C 1608 cm−1; νC = N 1585 cm−1.
7-Chloro-4-{(2E)-2-[1-(4-methylphenyl)ethylidene]hydrazinyl}quinoline (GPQF-8Q18)
Prepared in accordance with general synthetic procedure, which involved using 7-chloro-4-hydrazinylquinoline (1mmol) and 1-(4-methylphenyl)ethanone (1 mmol) as reactants, and a solution of ethanol and acetic acid 25% as the solvent. The reaction as maintained under reflux at 80 °C for 16 h. Yellow solid. Yield 41%. M.P. 310 °C (decomposition). 1H-NMR-(DMSO-d6)—δ = ppm: 2.38 (s, 3H, H22), 2.60 (s, 3H, H14), 7.32 (d, 2H, J = 6Hz, H18, H20), 7.51 (d, 1H, J = 9Hz, H3), 7.83 (d, 1H, J = 9Hz, H7), 7.90 (d, 2H, J = 6, H17, H21), 8.08 (s, 1H, H9), 8.82 (d, 1H, J = 9Hz, H2). IR—νNH 3466 cm−1; νCH 2555 cm−1; νC = C 1610 cm−1; νC = N 1585 cm−1.
7-Chloro-4-{(2E)-2-[1-(4-ethylphenyl)ethylidene]hydrazinyl}quinoline (GPQF-8Q19)
7-Chloro-4-hydrazinylquinoline (1mmol) and 1-(4-ethylphenyl)ethanone (1 mmol) were coupled in accordance with general synthetic procedure. Ethanol and acetic acid 25% were employed as the solvent, and the reaction was maintained under reflux at 80 °C for 16 h. Sharp yellow crystals. Yield 49%. M.P. 300–304 °C. 1H-NMR-(DMSO-d6)—δ = ppm: 1.22 (t, 3H, J = 9Hz, H23); 2.62 (s, 3H, H14); 2.68 (q, 2H, J = 6Hz, H22); 7.35 (d, 2H, J = 6Hz, H18, H20); 7,52 (d, 1H, J = 6Hz, H3); 7,85 (dd, 1H, J1 = 9Hz, J2 = 3Hz, H7); 7,92 (d, 2H, J = 6Hz, H17, H21); 8,13 (s, 1H, H9); 8,65 (d, 1H, J = 6Hz, H6); 8,86 (d, 1H, J = 9Hz, H2). IR—νNH 3473 e 3415 cm−1; νCH 2536 cm−1; νC = C 1608 cm−1; νC = N 1583 cm−1.
7-Chloro-4-{(2E)-2-[1-(3,4-dichlorophenyl)ethylidene]hydrazinyl}quinoline (GPQF-8Q20)
Prepared following the general synthetic procedure, which involved using 7-chloro-4-hydrazinylquinoline (1mmol) and 1-(3,4-dichlorophenyl)ethanone (1 mmol) as reactants, and a solution of ethanol/acetic acid 25%. The reaction was conducted under reflux at 80 °C for 16 h. Light orange solid. Yield 45%. M.P. 294–298 °C. 1H-NMR-(DMSO-d6)—δ = ppm 2.62 (s, 3H, H14); 7.57 (s, 1H, H17); 7.78 (d,1H, J = 9Hz, H21); 7.91 (d, 1H, J = 12Hz, H3); 7.99 (d, 1H, J = 6Hz, H20); 8.11 (s, 1H, H7); 8.21 (s, 1H, H9). IR—νNH 3473 and 3417 cm−1; νCH 2640cm−1; νC = C 1606 cm−1; νC = N 1587 cm−1.
7-Chloro-4-[(2E)-2-(4-phenylbutan-2-ylidene)hydrazinyl]quinoline (GPQF-8Q21)
Prepared in accordance with general synthetic procedure, which involved using 7-chloro-4-hydrazinylquinoline (1mmol) and 4-phenylbutan-2-one (1 mmol) as reactants, and a solution of ethanol/acetic acid 25% as the solvent. The reaction was maintained under reflux at 80 °C for 16 h. Beige solid. Yield 71%. M.P. 161–166 °C. 1H-NMR-(DMSO-d6)—δ = ppm: 2.21 (s, 3H, H23); 2.78 (t, 2H, J = 7.5 Hz, H15); 2.98 (t, 2H, J = 7.5 Hz, H14); 7.13 (d, 1H, J = 9 Hz, H3); 7.19–7.23 (m, 2H, H18, H20); 7.30–7.52 (m, 3H, H17, H19, H21); 7.77 (dd, 1H, J1 = 9 Hz, J2 = 3 Hz H7); 7.95 (s, 1H, H9); 8.53 (d, 1H, J = 9 Hz, H6); 8.64 (d, 1H, J = 9 Hz, H2). 13C-NMR-(75 MHz, DMSO-d6)—δ = ppm: 17.67 (C23); 31.03 (C15); 31.48 (C14); 100.18 (C3); 111.24 (C9); 118.14 (C5); 119.76 (C16); 125.97 (C19); 126.15 (C21); 126.52 (C17); 128.35 (C7); 128.39 (C6); 137.75 (C8); 139.78 (C13); 141.07 (C4); 143.54 (C10); 152.19 (C2). IR—νNH 3421cm−1; νCH 3055cm−1; νC = C 1606 cm−1; νC = N 1554 cm−1.
4-[(2E)-2-(butan-2-ylidene)hydrazinyl]-7-chloroquinoline (GPQF-8Q22)
Prepared using 7-chloro-4-hydrazinylquinoline (1mmol) and propan-2-one (1 mmol), in the presence of a solution of ethanol/acetic acid 25%, and left at room temperature for 24 h. Light yellow solid. Yield 77%. M.P. 71–74 °C. 1H-NMR-(CDCl3)—δ = ppm: 1.14 (t, 3H, J = 6Hz, H15); 2.09 (s,3H, H17); 2.33–2.41 (m, 2H, H14); 7.22 (d, 1H, J = 6Hz, H3); 7.51 (d, 1H, J = 9Hz, H7); 7.86 (s, 1H, H9); 8.37(d, 1H, J = 9Hz, H6); 8.53 (d, 1H, J = 6Hz, H2) 9.38 (s, 1H, H11). 13C-NMR-(75 MHz, DMSO-d6)—δ = ppm: 10.72 (C15); 14.56 (C17); 32.31 (C14); 102.12 (C3); 115.69 (C5); 120.39 (C9); 125.64 (C6 and C7); 134.90 (C8 and C13); 146.92 (C4); 161.88 (C10); 177.40 (C2). IR—νNH 3402 cm−1; νCH 2966 cm−1; νC = C 1610 cm−1; νC = N 1577 cm−1.
7-Chloro-4-[(2E)-2-(pentan-2-ylidene)hydrazinyl]quinoline (GPQF-8Q23)
Prepared using 7-chloro-4-hydrazinylquinoline (1mmol) and butan-2-one (1 mmol), in the presence of a solution of ethanol/acetic acid 25%. The reaction was mainteined at room temperature for 24 h. Yellow solid. Yield 51%. M.P. 250 °C (decomposition). 1H-NMR-(DMSO-d6)—δ = ppm: 0.96 (t, 3H, J = 7.5Hz, H16); 1.63–1.40 (m, 2H, H15); 2.21 (s, 3H, H18); 2.43 (t, 2H, J = 7.5Hz, H14); 7.24 (d, 1H, J = 6Hz, H3); 7.81 (d, 1H, J = 9Hz, H7); 8.12 (s, 1H, H9); 8.58 (d, 1H, J = 9Hz, H6); 8.81 (d,1H, J = 9Hz, H2). IR—νNH 3419 cm−1, νCH 2956–2648 cm−1; νC = C 1610 cm−1; νC = N 1587 cm−1.
7-Chloro-4-[(2E)-2-(3,3-dimethylbutan-2-ylidene)hydrazinyl]quinoline (GPQF-8Q25)
Prepared using 7-chloro-4-hydrazinylquinoline (1mmol) and 3,3-dimethybutan-2-one (1 mmol), with a solution of ethanol/acetic acid 25% as the solvent. The reaction was maintained at room temperature for 24 h. Light yellow solid. Yield 59%. M.P. 228–232 °C. 1H-NMR-(DMSO-d6)—δ = ppm: 1.23 (s, 9H, H19, H18, H15); 2.23 (s, 3H, H17); 7.27 (d, 1H, J = 9Hz, H7); 7.79 (d, 1H, J = 9Hz, H3); 8,17 (s, 1H, H9), 8.58 (d, 1H, J = 6Hz, H6); 8.83 (d, 1H, J = 9Hz, H2). 13C-NMR-(75 MHz, DMSO-d6)—δ = ppm: 13.78 (C17); 27.47 (C15, C18 and C19); 39.35 (C14); 100.18 (C3); 114.07 (C9); 119.10 (C5); 126.56 (C7); 126.69 (C6); 138.00 (C8); 139.15 (C13); 142.96 (C4); 152.80 (C10). IR—νNH 3419 cm−1; νCH 2970–2648 cm−1; νC = C 1608 cm−1; νC = N 1585 cm−1.
7-Chloro-4-{(2E)-2-[1-(4-chlorophenyl)ethylidene]hydrazinyl}quinoline (GPQF-8Q26)
7-Chloro-4-hydrazinylquinoline (1mmol) and 1-(4-chlorophenyl)ethanone (1 mmol) were coupled following the general synthetic procedure. A solution of ethanol/acetic acid 25% was used as the solvent, and the reaction was maintained under reflux at 80 °C for 16 h. Orange solid. Yield 67%. M.P. 290–292 °C. 1H-NMR-(DMSO-d6)—δ = ppm: 2.62 (s, 3H, H14); 7.58 (d, 3H, J = 9Hz, H3, H17, H21); 7.86 (d, 1H, J = 9Hz, H7); 8.03 (d, 2H, J = 9Hz, H18, H20); 8.08 (s, 1H, H9); 8.66 (d, 1H, J = 6Hz, H6); 8.82 (d, 1H, J = 9Hz, H2). IR—νNH 3481 and 3414cm−1; νCH 2607cm−1; νC = C 1622 cm−1; νC = N 1587 cm−1.
Biology
Animals, parasites, cells, and nematodes
The life cycle of S. mansoni (Belo Horizonte strain) is maintained through routine passage through Biomphalaria glabrata snails and Swiss mice at the Research Center on Neglected Diseases (Guarulhos University, SP, Brazil). Both rodents and snails were kept under environmentally controlled conditions (25 °C; humidity of 50%), with free access to food and water16. Vero cells were obtained from the American Type Culture Collection (ATCC CCL-81; Manassas, VA, USA). Cells were routinely cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 5% heat-inactivated fetal bovine serum and antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin) at 37 °C in a humidified atmosphere containing 5% CO2 17.
Caenorhabditis elegans wild-type (N2 strain), kindly provided by Dr. Carlos E. Winter (University of São Paulo), was routinely cultured at 22 °C on nematode growth medium (NGM) plates seeded with Escherichia coli (strain OP50) as a food source according to standard protocols 18.
In vitro antischistosomal assay
The antischistosomal assay was performed according to the methodology previously described16,19,20. Briefly, adult parasites, obtained from infected mice at day 42 post-infection, were incubated in flat bottom 24-well plates (Corning, New York, NY, USA) in RPMI 1640 culture medium (Vitrocell, Campinas, SP, Brazil) supplemented with 5% inactivated fetal calf serum, antibiotics (100 U/mL penicillin and 100 μg/mL streptomycin), and buffered with HEPES 25 mM (Sigma-Aldrich, St. Louis, MO, USA). Compounds were dissolved in dimethyl sulfoxide (DMSO at a final concentration of 0.5% v/v) and tested at 50 μM using PZQ 2 μM and DMSO 0.5% as positive and negative controls, respectively. Parasites were kept for 72 h (37 °C, 5% CO2, Panasonic, Osaka, Japan) and their viability was assessed using a Motic AE2000 inverted microscope (Vancouver, Canada)21. Each experiment was performed in triplicate and repeated at least three times. The negative control (using the highest concentration of DMSO, i.e., 0.5) and positive control (praziquantel 2 µM) were included.
For the determination of an effective concentration of 50% (EC50), compounds were tested using 1:2 serial dilutions from 50 to 3.12 µM as previously described22,23. Each concentration was tested in five replicates, and experiments were repeated once.
In vitro cytotoxicity assay
The cytotoxicity of the compounds on the Vero cells was determined by thiazolyl blue tetrazolium bromide (MTT) assay (Sigma-Aldrich) as previously described21,24. Briefly, cells were seeded using the density of 2 × 104 cells/well in a 96-well culture plate (Corning) and incubated with compounds (started at 200 µM and followed a 1:2 dilution series) at 37 °C and 5% CO2 (Panasonic). After 72 h, MTT solution was added to each well and the absorbance at 595 nm was read on a spectrophotometer (Epoch, BioTek Instruments, Winooski, VT, USA). The selectivity indices (SI) were calculated by dividing the 50% cytotoxic concentration (CC50) obtained on cells with 50% effective concentration (EC50) values determined on S. mansoni25.
In vivo antiparasitic studies in an animal model of schistosomiasis
In vivo studies in S. mansoni-infected mice were performed according to standard protocols20,26. Rodents (3 weeks old) were infected subcutaneously with 80 S. mansoni cercariae each and animals were randomly divided into experimental groups (five mice per group). Compounds were dissolved in 2% ethanol in water (v/v) and tested at a single oral dose (400 mg/kg) administered 42 days post-infection27. For comparison, praziquantel (400 mg/kg) and a corresponding amount of vehicle were administered to groups of five schistosomes-infected animals in the same period28,29. Two weeks after treatment, animals were placed in a chamber and euthanized by introducing 100% medical grade CO2 and therapeutic efficacy was based on the following aspects: (i) the number of schistosomes collected by portal perfusion (worm burden); (ii) quantitative fecal examination (egg burden determined by Kato-Katz technique), and (iii) eggs in tissue (egg burden in intestine determined by oogram method). as previously described30,31. The percentage of worm and egg reduction was calculated by means of the following equation: % reduction = [(value of untreated control group − value of treatment group)/value of untreated control group] × 100%32. The nonparametric Kruskal–Wallis test was used to compare the medians of the responses between the treatment and control groups using the GraphPad Prism software (version 8.0; CA, USA). Differences were considered statistically significant at P < 0.059.
In vitro toxicity assay in C. elegans model
For the drug assessment, the age-synchronized L1 stage was cultivated on standard NGM plates18. When at the L4 stage, approximately 25 nematodes were transferred to each well of a 96-wells NGM plates containing the drug of interest at the desired concentration (10, 25, 50, 100, and 200 μM) along with a positive control (levamisole 10 μM) and a negative control (DMSO 0.5%). Nematodes were kept at 22 °C for 24 h, and their viability was verified microscopically using a Motic AE2000 inverted microscope (Vancouver, Canada) equipped with an ultra-high definition (UHD) camera and with a 48-inch 4K-UHD monitor system (LG Electronics, São Paulo, Brazil)33. The worm viability was calculated by counting mobile worms and those considered dead which showed no movement on physical stimuli with a fine needle34.
Ethical approval
Animal studies are reported in compliance with the ARRIVE guidelines. The protocol for experimental design was reviewed and approved by the Committee for the Ethical Use of Animals in Experimentation at Guarulhos University, (Guarulhos, SP, Brazil; protocol ID 47/20) in accordance with the Guidelines for Care and Use of Laboratory Animals as stipulated by Brazilian laws.
Data availability
The data pertinent to this study is incorporated within the manuscript. The raw data supporting the findings can be made available by the corresponding author upon a reasonable request.
References
-
Santiago, E. D. F. et al. Evaluation of the anti-Schistosoma mansoni activity of thiosemicarbazones and thiazoles. Antimicrob. Agents Chemother. 58, 352–363 (2014).
Google Scholar
-
Fonseca, N. C. et al. Synthesis of a sugar-based thiosemicarbazone series and structure-activity relationship versus the parasite cysteine proteases rhodesain, cruzain, and Schistosoma mansoni cathepsin B1. Antimicrob. Agents Chemother. 59, 2666–2677 (2015).
Google Scholar
-
Correnti, J. M., Brindley, P. J. & Pearce, E. J. Long-term suppression of cathepsin B levels by RNA interference retards schistosome growth. Mol. Biochem. Parasitol. 143, 209–215 (2005).
Google Scholar
-
Gorka, A. P., de Dios, A. & Roepe, P. D. Quinoline drug-heme interactions and implications for antimalarial cytostatic versus cytocidal activities. J. Med. Chem. 56, 5231–5246 (2013).
Google Scholar
-
Liu, S. et al. Expression profile of the Schistosoma japonicum degradome reveals differential protease expression patterns and potential anti-schistosomal intervention targets. PLoS Comput. Biol. 10, e1003856 (2014).
Google Scholar
-
Hunt, P. R. & The, C. Elegans model in toxicity testing. J. Appl. Toxicol. 37, 50–59 (2017).
Google Scholar
-
Wu, T., Xu, H., Liang, X. & Tang, M. Caenorhabditis elegans as a complete model organism for biosafety assessments of nanoparticles. Chemosphere 221, 708–726 (2019).
Google Scholar
-
de Costa, D. S. et al. Antiparasitic properties of 4-nerolidylcatechol from Pothomorphe umbellata (L.) Miq. (Piperaceae) in vitro and in mice models with either prepatent or patent Schistosoma mansoni infections. J. Ethnopharmacol. 313, 116607 (2023).
Google Scholar
-
de Souza, R. L. et al. Enhancing the antischistosomal activity of carvacryl acetate using nanoemulsion. Nanomedicine 18, 331–342 (2023).
Google Scholar
-
Antinarelli, L. M. R. et al. Aminoquinoline compounds: Effect of 7-chloro-4-quinolinylhydrazone derivatives against Leishmania amazonensis. Exp. Parasitol. 171, 10–16 (2016).
Google Scholar
-
Bhat, H. R., Masih, A., Shakya, A., Ghosh, S. K. & Singh, U. P. Design, synthesis, anticancer, antibacterial, and antifungal evaluation of 4-aminoquinoline-1,3,5-triazine derivatives. J. Heterocycl. Chem. 57, 390–399 (2020).
Google Scholar
-
Kalita, J., Chetia, D. & Rudrapal, M. Design, synthesis, antimalarial activity and docking study of 7-chloro-4-(2-(substituted benzylidene)hydrazineyl)quinolines. Med. Chem. (Los. Angeles) 16, 928–937 (2020).
Google Scholar
-
Coimbra, E. S. et al. Amodiaquine analogs. Synthesis and anti-leishmanial activity. Mediterr. J. Chem. 1, 106–113 (2011).
Google Scholar
-
Candéa, A. L. P. et al. Synthesis and antitubercular activity of 7-chloro-4-quinolinylhydrazones derivatives. Bioorgan. Med. Chem. Lett. 19, 6272–6274 (2009).
Google Scholar
-
Montenegro, R. C. et al. 1-(7-Chloroquinolin-4-yl)-2-[(1H-pyrrol-2-yl)methylene]hydrazine: A potent compound against cancer. Med. Chem. Res. 21, 3615–3619 (2012).
Google Scholar
-
Brito, J. R. et al. Neolignans isolated from Saururus cernuus L. (Saururaceae) exhibit efficacy against Schistosoma mansoni. Sci. Rep. 12, 19320 (2022).
Google Scholar
-
Silva, T. C. et al. N-(4-Methoxyphenyl)pentanamide, a simplified derivative of albendazole, displays anthelmintic properties against the nematode Toxocara canis. Microbiol. Spectr. 10, 13 (2022).
-
Stiernagle, T. Maintenance of C. elegans. WormBook 11, 1–11 (2006).
-
de Moraes, J., Dario, B. S., Couto, R. A. A., Pinto, P. L. S. & da Costa Ferreira, A. M. Antischistosomal activity of oxindolimine-metal complexes. Antimicrob. Agents Chemother. 59, 6648–6652 (2015).
Google Scholar
-
Lago, E. M. et al. Phenotypic screening of nonsteroidal anti-inflammatory drugs identified mefenamic acid as a drug for the treatment of schistosomiasis. EBioMedicine 43, 370–379 (2019).
Google Scholar
-
Sessa, D. P. et al. 15β-Senecioyl-oxy- ent -kaur-16-en-19-oic acid, a diterpene isolated from Baccharis lateralis, as promising oral compound for the treatment of schistosomiasis. J. Nat. Prod. 83, 3744–3750 (2020).
Google Scholar
-
de Brito, M. G. et al. Therapeutic effect of diminazene aceturate on parasitic blood fluke Schistosoma mansoni infection. Antimicrob. Agents Chemother. 64, 33 (2020).
-
Porto, R. et al. Antiparasitic properties of cardiovascular agents against human intravascular parasite Schistosoma mansoni. Pharmaceuticals 14, 1–15 (2021).
Google Scholar
-
Dematei, A. et al. Mechanistic insights into the leishmanicidal and bactericidal activities of batroxicidin, a cathelicidin-related peptide from a South American Viper (Bothrops atrox). J. Nat. Prod. 84, 1787–1798 (2021).
Google Scholar
-
Morais, C. S. et al. Pyrazoline derivatives as promising novel antischistosomal agents. Sci. Rep. 11, 23437 (2021).
Google Scholar
-
Xavier, R. P. et al. H1-antihistamines as antischistosomal drugs: in vitro and in vivo studies. Parasit. Vectors 13, 278 (2020).
Google Scholar
-
Silva, M. P. et al. Brazilian red propolis exhibits antiparasitic properties in vitro and reduces worm burden and egg production in an mouse model harboring either early or chronic Schistosoma mansoni infection. J. Ethnopharmacol. 264, 113387 (2021).
Google Scholar
-
Mengarda, A. C. et al. Antiparasitic activity of piplartine (piperlongumine) in a mouse model of schistosomiasis. Acta Trop. 105350 (2020). https://doi.org/10.1016/j.actatropica.2020.105350.
-
Mengarda, A. C. et al. Licarin A, a neolignan isolated from Nectandra oppositifolia Nees & Mart. (Lauraceae), exhibited moderate preclinical efficacy against Schistosoma mansoni infection. Phyther. Res. 35, 5154–5162 (2021).
-
Roquini, D. B. et al. Promethazine exhibits antiparasitic properties in vitro and reduces worm burden, egg production, hepatomegaly, and splenomegaly in a schistosomiasis animal model. Antimicrob. Agents Chemother. 63, 12 (2019).
-
Guerra, R. A. et al. In vitro and in vivo studies of spironolactone as an antischistosomal drug capable of clinical repurposing. Antimicrob. Agents Chemother. 63, 1722 (2019).
-
Xavier, E. S. et al. Therapeutic efficacy of carvacrol-loaded nanoemulsion in a mouse model of schistosomiasis. Front. Pharmacol. 13, 917363 (2022).
-
Rocha, V. C. et al. Evaluating the antischistosomal activity of dehydrodieugenol B and its methyl ether isolated from Nectandra leucantha—A preclinical study against Schistosoma mansoni infection. ACS Omega 8, 40890–40897 (2023).
Google Scholar
-
Roquini, D. B. et al. Susceptibility of Angiostrongylus cantonensis larvae to anthelmintic drugs. Front. Pharmacol. 13, 901459 (2022).
Acknowledgements
The authors express gratitude to Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP 2019/26686-2) for the financial support and to Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES 88887.505029/2020-00) for the scholarships provided to T.F.A.P. This study received partial funding from CAPES (Finance Code 001) for M.E.C. and T.R.T., and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, grant #312211/2021-0 to J.d.M). The authors acknowledge Mariana B. Silva for her excellent technical assistance at the Research Center on Neglected Diseases (Guarulhos University, Brazil).
Author information
Authors and Affiliations
Contributions
D.G.G.R. and T.F.A.P. carried out the conceptualization of the study. T.F.A.P. performed the synthesis and characterization of the compounds. M.E.C. and T.R.T. performed the biological assays. D.G.G.R., T.F.A.P., M.E.C., T.R.T. and J.d.M. carried out the formal analysis and data curation. D.G.G.R and J.d.M contributed with resources. T.F.A.P. and D.G.G.R. wrote original draft and edited the final version. D.G.G.R., T.F.A.P., M.E.C., T.R.T. and J.d.M. reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Supplementary Figures.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Reprints and Permissions
About this article
Cite this article
Pavani, T.F.A., Cirino, M.E., Teixeira, T.R. et al. Targeting the Schistosoma mansoni nutritional mechanisms to design new antischistosomal compounds.
Sci Rep 13, 19735 (2023). https://doi.org/10.1038/s41598-023-46959-3
-
Received: 27 September 2023
-
Accepted: 07 November 2023
-
Published: 13 November 2023
-
DOI: https://doi.org/10.1038/s41598-023-46959-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.