Abstract
A new sensitive method to determine polonium-210 (210Po) and lead-210 (210Pb) in a diversity of environmental samples was developed. For fresh and marine waters, Po was pre-concentrated using a titanium (III) hydroxide (Ti(OH)3) co-precipitation. Solid environmental samples were digested with nitric acid (HNO3) and hydrogen peroxide (H2O2). The alpha thin layer source was prepared using CuS micro-precipitation and 210Po was measured by alpha spectrometry. Lead-210 was left to decay for up to a year and indirectly measured via its progeny, 210Po. The chemical recoveries for 210Po and 210Pb were high, 90% and 97%, respectively, for a large variety of samples and a very low minimum detectable activity (MDA) was obtained. The method was validated using standardized solutions and certified reference materials.
Introduction
Polonium-210 (210Po) is a naturally occurring alpha-emitting radioisotope from the uranium-238 (238U) decay series with a high activity per unit mass (1.66 × 1014 Bq g−1) and a relatively short radioactive half-life (t1/2 = 138.4 days)1, making it one of the most toxic radionuclides in the environment2,3,4. Lead-210 (210Pb) (t1/2 = 22.2 years)1 is a beta-emitting radioisotope that decays to bismuth-210 (210Bi) (t1/2 = 5.0 days)1, and then to 210Po. Lead-210 and 210Po are linked by their radiogenic association and can be in secular equilibrium in static environments or unsupported by each other in dynamic conditions2,4,5. Naturally, 210Po and 210Pb activities could be more elevated in some regions based on environmental characteristics such as local geology, land cover or forest fires6. These radionuclides can also be anthropogenically enriched by activities such as mining, fracking and fossil fuel combustion, which bring 238U and its decay products to the earth’s surface and surrounding environments7,8,9.
Despite the high toxicity and significant contribution of 210Po to the natural radiation dose received by living organisms (> 90%)10,11, 210Po remains the least studied natural radionuclide in freshwater environments5,12,13. This is mainly because some of its compounds are volatile and there are limited rapid methods to measure it in low-level environmental samples containing a high amount of organic matter such as animal tissues, foodstuff and vegetation12,14. Traditionally, 210Po has been determined using the spontaneous deposition technique where 210Po is self-plated on a metallic disc in an acidic solution13,15. This method while effective for some applications, has limitations for us in large-scale environmental studies. These limitations include high sample throughput times, detection limits not suitable for all environmental samples and the possibility of interference of ions, such as iron and chromium, with the Po plating16,17,18. Conversely, 210Pb has been well studied in several environmental samples19,20. Measurements of 210Pb can be efficiently determined by gamma spectrometry in solid sample matrices if a large amount of sample is available and/or if the activity concentrations are high enough. However, the determination of trace activities of 210Pb in certain matrices and small size samples, such as water and freshwater organisms, can be labour intensive, expensive and sometimes impossible due to detection limits that are too high21.
Without adequate methods to measure environmental levels of 210Pb and 210Po, there is a significant lack of data on the occurrence of these radionuclides in the environment; thus, insufficient data are available to understand their behaviour in the environment The current lack of understanding on the activities and movement of 210Pb and 210Po in the environment, combined with increased industrial activities that can elevate their concentrations and mobility in ecosystems such as uranium mining; as well as natural events, such as forest fires underscores the importance of developing a simple and rapid method to determine these radionuclides to support robust and reliable environmental studies and thereby generate more accurate risk assessments for human and non-human biota11,14.
A more efficient and accurate method was developed by Guérin and Dai22,23 to prepare thin alpha layer sources of Po using copper sulfide (CuS) micro-precipitation. This method consists of selectively co-precipitating polonium sulfide (PoS) with CuS from other potential interferences in an acidic solution. This method had only been used for simple matrices such as drinking water and not for more complex environmental samples, such as biological tissues. To expand the applicability of this method, a novel method to determine 210Po and, indirectly, 210Pb, in a variety of abiotic (fresh and marine waters) and biotic (freshwater organisms and vegetation) environmental samples is presented. The recovery was optimized, the MDA was determined, and the method was validated.
Materials and methods
Reagents and standards
All chemicals used were analytical grade except acids, which were trace metal grade. Ultrapure water (UPW) from a Milli-Q water system (Millipore, Bedford MA, USA) was used for the preparation of all reagents and standards. Hydrochloric acid (HCl), nitric acid (HNO3), hydrogen peroxide (H2O2), sodium hydroxide (NaOH), copper chloride (CuCl2·2H2O), and ethanol (CH3CH2OH) were purchased from Fisher Scientific (Fair Lawn, NJ). Titanium trichloride (TiCl3) and sodium sulfide (Na2S·9H2O) were obtained from Millipore Sigma (St. Louis, MO).
Certified standard solutions of 210Po, 209Po and 210Pb were purchased from Eckert & Ziegler Isotope Products (Valencia, CA). In addition, reference materials (CLV1 and CLV2) containing 210Po and 210Pb, in equilibrium, were purchased from the International Atomic Energy Agency (IAEA; Vienna, Austria)24. The accuracy of the certified reference materials was tested by an inter-laboratory program established by Natural Resources Canada25. A certified stable lead standard was purchased from Millipore Sigma (Oakville, ON).
Analytical equipment
Radiological activity concentrations (209Po, 210Po and 210Pb) were detected using an Alpha Analyst alpha spectrometer (Mirion Technologies (Canberra), Meriden, CT) with a counting efficiency superior to 17% and a background of < 1 count/hour in the energy regions of interest. Apex Alpha counting productivity software was used for detector setup and control, quality assurance, sample analysis and elaboration. The Pb chemical yield was verified by inductively coupled mass plasma spectrometry (ICP-MS) (8900 ICP-MS Triple Quad, Agilent Technologies, Santa Clara, CA).
Sample collection and preservation
Surface water
To test the applicability of this method for a range of water types, surface waters were collected from marine (n = 4) and fresh water (n = 13) systems representing a gradient of ionic content (Supplementary Fig. S1, Supplementary Fig S2 and Supplementary Table S1). Marine surface waters (4 L) were collected within the St. Lawrence Estuary and Gulf of St. Lawrence in eastern Canada. Freshwater samples (4 L) were collected from Perch Lake in Ontario,Canada located on the Chalk River Laboratories (CRL) site. Immediately following collection, samples were filtered using a 0.45 µm filter (FHT-45, Waterra, Mississauga, ON) and acidified to 1% HNO3. The electrical conductivity of the samples was measured using an Oakton 450 Meter (Thomas Scientific, Swedesboro, NJ), and the elemental content was measured by ICP-MS (Supplementary Table S2). The range of conductivity, calcium mass concentration, and chloride mass concentration for the samples collected are shown in Table 1.
Biota
Biota from selected trophic levels were collected in the Lac Granet and Lac Camille-Roy systems (main lake and connected streams) of western Québec, Canada. Biotic samples collected included algae (n = 3), macrophytes (n = 7), plankton (n = 2), invertebrates (n = 9), molluscs (n = 6) and fish (n = 18). Plankton samples were collected with plankton nets (WildCo, Saginaw, MI) mesh sizes 153 µm and towed behind a boat. Algae, macrophytes, invertebrates and fish were collected using dip and kick nets (WildCo, Saginaw, MI). Immediately after collection, invertebrates and molluscs were placed in UPW and allowed to purge their gut contents for approximately 12 hours26. All samples were thoroughly rinsed in lake water and kept frozen until further processing.
Procedure
Sample processing and digestion
The samples were processed within 30 days of collection to reduce 210Po loss from radioactive decay and 210Po ingrowth from 210Pb. For the water samples, a one litre subsample was weighed into a glass beaker and acidified to pH 2 using concentrated HNO3 to stabilize the sample and keep the Po in solution. Next, a known amount of 209Po yield tracer (approximately 15 mBq) and stable Pb standard (approximately 2.0 µg) was added. The sample was filtered again through a 0.22 µm polystyrene filter (Fisher Scientific, Fair Lawn, NJ) to remove remaining particulate matter and colloids.
Biota samples, were processed wet and were minced using a scalpel and scissors and homogenized before distributing 1 to 2 g (wet weight) into a 15 mL polypropylene digestion tube. A known amount of 209Po (approximately 15 mBq) was added to each sample before digestion. To digest the sample, 3.3 mL of concentrated HNO3 were added to the digestion tube and allowed to sit for approximately 1 h followed by the addition of 2.0 mL of 30% H2O2. The sample solution was then heated to 50 °C for 48 h. Once removed, the dissolved sample solution was allowed to cool and it was transferred to a 50 mL polypropylene conical tube (Cole-Parmer, Montreal, QC). The sample was centrifuged at 3500 rpm for 1.5 min. The supernate was transferred to a new 15 mL polypropylene conical tube (Cole-Parmer, Montreal, QC) and diluted to a volume of 10 mL with UPW.
Water samples pre-concentration
For the water samples only, Po was co-precipitated with Ti(OH)3 using 0.6 mL of 10% m v−1 TiCl3, and the pH was adjusted between 10 and 12 using about 30 mL of 40% mv−1 sodium hydroxide (NaOH) solution. The fresh and marine water samples were left aside for 1 and 4 h, respectively, which allowed the Ti(OH)3 to settle and to easily remove most of the supernate by decantation. The precipitate was then recovered by centrifugation, dissolved with 3.3 mL of concentrated HNO3 and diluted to a volume of 10 mL with UPW.
CuS microprecipitation
. The 10 mL, 5 mol l−1 HNO3, supernate collected following the water pre-concentration and biota sample digestion, was filtered through a 0.1 µm Resolve™ filter (Eichrom Technologies Inc., Lisle, IL) using a multi-hole vacuum box and recovered in a 50 mL polypropylene conical tube. Then, 0.2 mL of 500 µg mL−1 Cu(II) solution was added and mixed, followed immediately by the addition of 0.2 mL of 10% m v−1 Na2S, at which point a visible (brown) colloidal precipitate formed. The sample was then filtered immediately through a 0.1 µm Resolve™ filter. After rinsing the filter with 1 to 2 mL of UPW, the filtrate was set aside for future 210Pb determination and replaced with a 50 mL polypropylene conical waste tube. The filter was rinsed with 1 to 2 mL of 80% ethanol. The precipitate retained on the filter was air-dried and mounted on a stainless steel disc (AF Murphy Die & Machine Co Inc., North Quincy, MA) for 210Po determination by alpha spectrometry. Water samples were counted for 48–96 h, while biota samples were counted for 24 to 48 h. The in situ 210Po activity concentrations were calculated by Eq. (1):
where A’Po-210 is the 210Po activity at the time of extraction, A’Po-210 ingrowth is a correction factor for 210Po loss as well as 210Po ingrowth from 210Pb decay from the time of sampling and extraction, λPo is the decay constant for 210Po and T is time between sample collection and extraction27,28.
210Pb determination
Lead-210 activity concentrations were determined by measuring the ingrowth activity of its daughter, 210Po, using alpha spectrometry. Following the CuS micro-precipitation step, an additional aliquot of approximately 15 mBq of 209Po yield tracer and 2.0 µg of stable Pb carrier was added to the final filtrate. The sample was then left aside for at least 4 months to allow210Po ingrowth. Finally, the CuS micro-precipitation was repeated a second time and the 210Pb activity was calculated using Eq. (2):
where APb-210 in situ is the activity concentration of the sample at the time of collection, AmPo is the activity of 210Po at the time of the second CuS micro-precipitation, t2 is a correction factor for the decay of 210Pb from the time of sample collection and second extraction, t1 is a ingrowth factor for 210Po from the decay of 210Pb for time between first and second extraction, λPo and λPb are decay constants for 210Po and 210Pb, respectively27,28. The nc parameter is the Pb chemical yield calculated by applying the following Eq. (3):
where CPb is the lead concentration detected by ICP-MS; V1 is the volume collected from the filtrate; CPbcarrier is the concentration of the Pb carrier added; and V2 is the initial sample volume.
Method development
The method was applied to a variety of environmental samples, which included different matrices and sample sizes to tests its applicability for large scale field studies. For water samples, an additional filtration step was added before the micro-precipitation step to improve the chemical resocvery. In addition, following the Ti(OH)3 co-precipitation of marine waters, a longer settling period was essential to improve accuracy. To assess the possible loss of Pb during the Ti(OH)3 co-precipitation and CuS micro-precipitation steps, a stable lead standard was added and an aliquot was taken before and after the extraction.
Figures of merit
The MDA was determined by measuring 210Po and 210Pb in ten reagent blank samples following all the steps of the described method and using the Currie equation28 (4):
where k is a constant (1.645) to reach the 95% confidence interval, B is the number of background counts for a defined time in seconds (T), ε is the counting efficiency, R is the chemical recovery, V is the volume in litres and F is a unit conversion factor which equals 10−3.
The method accuracy and precision were determined by measuring 210Po (n = 7) and 210Pb (n = 3) in water samples spiked with a known activity of 210Po and210Pb, in equilibrium, using the developed method. The relative bias (Bri) and the relative precision (SB) were calculated using Eqs. 5 and 6. 29,30:
where Ai is the measured activity, Aai is the added activity, Br is the mean relative bias and N is the number of replicates.
The method was validated for solid samples by determining the 210Po and 210Pb activity concentration in certified reference materials CLV-1 and CLV-2. The data followed a normal distribution and therefore a Student’s t-test was used to evaluate differences in the 210Po and 210Pb activity concentrations measured in the certified reference materials by the inter-laboratory program and this method.
Animal care
The institutional review board that reviewed and approved the capture, handling and euthanasia of fish for this study was the Wildlife Management Branch of the province of Québec’s Ministry of Forests, Wildlife and Parks (permit # 2021-07-14-070-08-GP). This approved and issued permit covered all aspects of fish collection, animal welfare and euthanasia methods. All methods were carried out in accordance with relevant guidelines and regulations. Euthanasia of fish was conducted following Guideline 113 of the Canadian Council on Animal Care guidelines on the care and use of fish in research, teaching and testing31. Specifically, fish euthanasia was conducted immediately after capture by swift blow to the head (destruction of brain tissue). Where relevant, all methods are reported in accordance with ARRIVE guidelines (https://arriveguidelines.org) for the reporting of animal experiments.
Results and discussion
Recovery optimization
Initially, a chemical recovery of 37 ± 10% was obtained for environmental fresh water samples, which was much lower than test samples prepared with UPW (~ 85%). The average recovery increased to 89.8 ± 12.5% when the samples were re-filtered before the CuS micro-precipitation step using a 0.22 µm polyethersulfone filter (Fisher Scientific, Fair Lawn, NJ) to remove suspended particulate matter and colloids smaller than 0.45 µm in size. During the final filtration step, following the CuS micro-precipitation, the PoS precipitate as well as any particulate matter would be retained leading to a poor spectral resolution and recovery. This additional filtration step substantially improved the chemical yield and spectral resolution for field water samples.
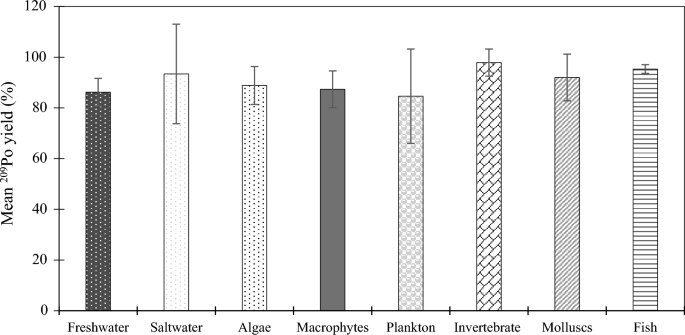
Next, the method was tested on various types of samples and the results are shown in Fig. 1. The chemical recovery was high for all type of samples tested (90.7 ± 9.3%), which demonstrated that this method was versatile and robust.

209Po recovery for initial 210Po extraction as a function of sample type. Error bars indicate the standard error of the mean.
Approximately 4 months after the initial 210Po extractions, the same samples were analyzed again to indirectly measure the 210Pb present in the sample. The chemical recovery following the ingrowth period was high (97.1 ± 8.2%) and consistent for all sample types as shown in Fig. 2.

209Po recovery as a function of sample type for indirect 210Pb measurement via 210Po extraction following a 4 month in growth period. Error bars indicate the standard error of the mean.
Figures of merit
A very low MDA of 0.05, 0.02, and 0.01 mBq L−1 was obtained for 24, 48, and 96 h of counting, respectively (Table 2) The method was validated for water samples using water samples spiked with 210Po and 210Pb with a range of activities from 12 to 160 mBq and counted for 24, 48 and 96 h (Fig. 3). The mean 210Po chemical recovery was 97 ± 7% over the 3 count times. Three samples were allowed to sit for approximately 4 months when the CuS micro-precipitation was repeated to validate the method for 210Pb determination (Fig. 4) (mean 210Pb chemical recovery was 102 ± 7% and mean stable Pb chemical recovery was 108 ± 6%). To further validate the method for solid samples, two certified reference materials (CLV-1 and CLV-2) with a range of 210Po and 210Pb activities from 74 to 660 mBq g−1 were chosen to represent a range of activity concentrations. Mean 210Po chemical recoveries of 89 ± 3% and 80 ± 6% were calculated from the analyses of the two vegetation reference materials CLV-1 (n = 5) and CLV-2 (n = 4), respectively. A mean 210Pb chemical recovery of 101 ± 9. % was calculated from the analyses of the vegetation reference material CLV-1 (n = 5) (Table 2 and Figs. 5, 6). Furthermore, the comparison of the expected and measured 210Po and 210Pb activities in reference materials using a two-tailed Student’s t-test, assuming unequal variances (p-values > 0.05), indicates repeatability, confirming the method to be accurate for 210Po and 210Pb determination in environmental abiotic and biotic non-water samples. These results provided evidence that complex matrices, as well as reduced sample size and digestion temperatures, produced consistent and high polonium and lead recoveries (78–101%) (Table 3). The developed method was also accurate and precise as evidenced by the mean relative bias and mean relative precision presented in Table 2.

210Po recovery results for spiked water samples after CuS micro-precipitation.

210Pb recovery results for spiked water samples following a 4 month in-growth period.

210Po recovery results for certified vegetation standards after CuS micro-precipitation.

210Pb recovery results for certified vegetation standards after CuS micro-precipitation.
Application and comparison of the method
The mean activity concentrations of 210Po measured in different types of waters (freshwater and saltwater) and several types of freshwater organisms are shown in Fig. 7. Freshwater organism samples such as periphyton and invertebrates are often too small in mass to be able to accurately measure their 210Po activity concentration. For example, sample size requirements and detection limits for other methods in the literature are summarized in Table 4. The sample quantity requirements and/or detection limits for these methods are not applicable to many environmental sample types. However, the developed method was successful in reducing sample quantities while maintaining sufficiently sensitive detection limits (Fig. 8). Furthermore, applying the CuS microprecipitation method to indirectly measure 210Pb not only provided a more sensitive technique, it also allowed the same sample to be used for both the 210Po and 210Pb analyses, which reduced field sampling efforts and laboratory processing times.

Mean 210Po activity concentrations measured for water samples and solid samples. Error bars indicate the standard error of the mean.

Mean MDAs measured for water samples and solid samples. Error bars indicate the standard error of the mean.
Conclusion
Large data gaps exist in understanding the behavior of the 210Po and 210Pb in the environment, and this is largely due to challenges associated with measuring these radionuclides at the low-level activity concentrations typically encountered in the environment14,18. The developed method is simple and highly sensitive (MDA ~ 0.01 mBq L−1). Consistent and high chemical recoveries (> 80%) for abiotic and biotic environmental samples were obtained, thereby making it possible to adequately study the behavior of 210Po and 210Pb in the environment.
Data availability
The 210Po and 210Pb dataset generated during this research is being prepared for a separate manuscript as part of a doctoral thesis. The datasets generated during the current study are available from the corresponding author on reasonable request.
References
-
Laboratory National Henri Becquerel (LNHB). Atomic and Nuclear Data. http://www.lnhb.fr/nuclear-data/nuclear-data-table/ (2022).
-
Scott, B. R. Health risk evaluations for ingestion exposure of humans to Polonium-210. Dose Response 5, 94–122. https://doi.org/10.2203/dose-response.06-013.Scott (2007).
Google Scholar
-
Ansoborlo, E. et al. Review of chemical and radiotoxicological properties of polonium for internal contamination purposes. Chem. Res. Toxicol. 25, 1551–1564. https://doi.org/10.1021/tx300072w (2012).
Google Scholar
-
Carvalho, F. et al. The environmental behaviour of polonium. Int. At. Energy Agency Tech. Rep. Ser. 484, 1–255 (2017).
-
International Commission on Radiological Protection (ICRP). ICRP Publication Annual Report 137, 1–22, ICRP 2017 Annual Report.pdf (2017).
-
Carvalho, F. P., Oliveira, J. M. & Malta, M. Exposure to radionuclides in smoke from vegetation fires. Sci. Total Environ. 472, 421–424. https://doi.org/10.1016/j.scitotenv.2013.11.073 (2014).
Google Scholar
-
Clulow, F. V., Dave, N. K., Lim, T. P. & Avadhanula, R. Radium-226 in water, sediments, and fish from lakes near the city of Elliot Lake, Ontario, Canada. Environ. Pollut. 99, 199–213. https://doi.org/10.1016/S0269-7491(97)00176-0 (1998).
Google Scholar
-
Warner, N. R., Christie, C. A., Jackson, R. B. & Vengosh, A. Impacts of shale gas wastewater disposal on water quality in Western Pennsylvania. Environ. Sci. Technol. 47, 11849–11857. https://doi.org/10.1021/es402165b (2013).
Google Scholar
-
Lauer, N., Vengosh, A. & Dai, S. Naturally occurring radioactive materials in uranium-rich coals and associated coal combustion residues from China. Environ. Sci. Technol. 51, 13487–13493. https://doi.org/10.1021/acs.est.7b03473 (2017).
Google Scholar
-
Cherry, R. D. & Heyraud, M. Evidence of high natural radiation doses in certain mid-water oceanic organisms. Science 218, 54–56. https://doi.org/10.1126/science.7123217 (1982).
Google Scholar
-
Thakur, P. & Ward, A. L. 210Po in the environment: Insight into the naturally occurring polonium isotope. J. Radioanal. Nucl. 323, 27–49. https://doi.org/10.1007/s10967-019-06939-2 (2020).
Google Scholar
-
Henricsson, F., Ranebo, Y., Holm, E. & Roos, P. Aspects on the analysis of 210Po. J. Environ. Radioact. 102, 415–419. https://doi.org/10.1016/j.jenvrad.2010.09.010 (2011).
Google Scholar
-
Fonollosa, E., Peñalver, A., Aguilar, C. & Borrull, F. Polonium-210 levels in different environmental samples. Environ. Sci. Pollut. Res. 22, 20032–20040. https://doi.org/10.1007/s11356-015-5158-3 (2015).
Google Scholar
-
Matthews, K. M., Kim, C. K. & Martin, P. Determination of 210Po in environmental materials: A review of analytical methodology. Appl. Radiat. Isot. 65, 267–278. https://doi.org/10.1016/j.apradiso.2006.09.005 (2007).
Google Scholar
-
Sethy, N. K. et al. A review of radio chemical analysis and estimation of 210Po in soil matrices. J. Radiat. Res. Appl. Sci. 8, 590–596. https://doi.org/10.1016/j.jrras.2015.07.001 (2015).
Google Scholar
-
Wilkinson, P. The determination of environmental levels of uranium and thorium series isotopes and 137Cs in aquatic and terrestrial samples. Can. J. Fish Aquat. 78, 1–51 (1985).
-
Seiner, B. E. et al. Effects of digestion, chemical separation and deposition on Po-210 quantitative analysis. J. Radioanal. Nucl. 302, 673–678. https://doi.org/10.1007/s10967-014-3255-2 (2014).
Google Scholar
-
Kong, X., Yin, L. & Ji, Y. Simultaneous determination of 210Pb and 210Po in seafood samples using liquid scintillation counting. J. Environ. Radioact. 231, 1–6. https://doi.org/10.1016/j.jenvrad.2021.106553 (2021).
Google Scholar
-
Vesterbacka, P. & Ikaheimonen, T. K. Optimization of 210Pb determination via spontaneous deposition of 210Po on a silver disk. Anal. Chim. Acta 546, 252–261. https://doi.org/10.1016/j.aca.2005.04.074 (2005).
Google Scholar
-
Ebaid, Y. & Khater, A. E. M. Determination of 210Pb in environmental samples. Radioanal. Nucl. Chem. 270, 609–619. https://doi.org/10.1007/s10967-006-0470-5 (2006).
Google Scholar
-
García-Orellana, I. & García-León, M. An easy method to determine Po-210 and Pb-210 by alpha spectrometry in marine environmental samples. Appl. Radiat. Isot. 56, 633–636. https://doi.org/10.1016/S0969-8043(01)00261-5 (2002).
Google Scholar
-
Guérin, N. & Dai, X. H. Rapid preparation of polonium counting sources for alpha spectrometry using copper sulfide microprecipitation. Anal. Chem. 85, 6524–6529. https://doi.org/10.1021/ac401311b (2013).
Google Scholar
-
Guérin, N. & Dai, X. H. Determination of 210Po in drinking water and urine samples using copper sulfide microprecipitation. Anal. Chem. 86, 6026–6031. https://doi.org/10.1021/ac501164f (2014).
Google Scholar
-
Natural Resources Canada (NRCAN). CLV-1 and CLV-2 Certificate of Analysis, https://www.nrcan.gc.ca/our-natural-resources/minerals-mining/mining-resources/clv-1-and-clv-2-certificate-analysis/8079 (2022).
-
Dalton, J.L. & Bowman, W.S. National Uranium Tailings Program, report: NUTP-4E, 1–20. (1986).
-
Gillis, P. L., Chow-Fraser, P., Ranville, J. F., Ross, P. E. & Wood, C. M. Daphnia need to be gut-cleared too: The effect of exposure to and ingestion of metal-contaminated sediment on the gut-clearance patterns of D. magna. Aquat. Toxicol. 71, 143–154. https://doi.org/10.1016/j.aquatox.2004.10.016 (2005).
Google Scholar
-
Baskaran, M. et al. Effects of flow rates and composition of the filter, and decay/ingrowth correction factors involved with the determination of in situ particulate 210Po and 210Pb in seawater. Limnol. Oceanogr. Methods 11, 126–138. https://doi.org/10.4319/lom.2013.11.126 (2013).
Google Scholar
-
Currie, L. A. Limits for qualitative detection and quantitative determination. Application to radiochemistry. Anal. Chem. 40, 586–593. https://doi.org/10.1021/ac60259a007 (1968).
Google Scholar
-
Walther, B. A. & Moore, J. L. The concepts of bias, precision and accuracy, and their use in testing the performance of species richness estimators, with a literature review of estimator performance. Ecography 28, 815–829. https://doi.org/10.1111/j.2005.0906-7590.04112.x (2005).
Google Scholar
-
Burton, A., Altman, D. G., Royston, P. & Holder, R. L. The design of simulation studies in medical statistics. Stat. Med. 25, 4279–4292. https://doi.org/10.1002/sim.2673 (2006).
Google Scholar
-
Canadian Council on Animal Care (CCAC). Guideline on: The care and use of fish in research, teaching and testing. https://ccac.ca/Documents/Standards/Guidelines/Fish.pdf. (2005).
-
International Atomic Energy Agency (IAEA). A procedure for the determination of Po-210 in water samples by alpha spectrometry. IAEA Anal. Qual. Nucl. Appl. Ser. 12, 1–42 (2009).
-
Biggin, C. C., Cook, G., Mackenzie, A. B. & Pates, J. M. Time efficient method for determind of 210Pb, 210Bi and 210Po activity in seawater using liquid scintillation spectrometry. Anal. Chem. 74, 671–677. https://doi.org/10.1021/ac0107599 (2002).
Google Scholar
-
Sadi, B. B., Chen, J., Kochermin, V., Tung, G. & Chiorean, S. A faster sample preparation method for determination of polonium-210 in fish. J. Radioanal. Nucl. Chem. 308, 843–850. https://doi.org/10.1007/s10967-015-4575-6 (2016).
Google Scholar
-
Khan, M. F., Wesley, S. G. & Rajan, M. P. Polonium-210 in marine mussels (bivalve molluscs) inhabiting the southern coast of India. J. Environ. Radioact. 138, 410–416. https://doi.org/10.1016/j.jenvrad.2014.06.023 (2014).
Google Scholar
Acknowledgements
This work was funded by the AECL Federal Nuclear Science and Technology Work Plan. The research was designed and conducted at the Canadian Nuclear Laboratories. The authors would like to acknowledge Hailly Degage for assisting with sample collection and sample processing, Dan Festarini for preparing maps of the sampling locations, as well as Emily Saurette, Elizabeth Priebe and Lars Brinkmann for their comments and support.
Author information
Authors and Affiliations
Contributions
S.W., M.B. and D.J.R. designed the research and collected the environmental samples. S.W. performed the sample preparations and measurements of 210Po and 210Pb. J.B provided helpful suggestions for the research. S.W., M.B. and N.G. wrote the manuscript. All authors discussed the results and contributed to the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Supplementary Information.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Reprints and Permissions
About this article
Cite this article
Walsh, S., Bond, M.J., Guérin, N. et al. A sensitive method to determine 210Po and 210Pb in environmental samples by alpha spectrometry using CuS micro-precipitation.
Sci Rep 13, 19754 (2023). https://doi.org/10.1038/s41598-023-46230-9
-
Received: 02 March 2023
-
Accepted: 30 October 2023
-
Published: 13 November 2023
-
DOI: https://doi.org/10.1038/s41598-023-46230-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.