Abstract
Natural language-based generative artificial intelligence (AI) has become increasingly prevalent in scientific research. Intriguingly, capabilities of generative pre-trained transformer (GPT) language models beyond the scope of natural language tasks have recently been identified. Here we explored how GPT-4 might be able to perform rudimentary structural biology modeling. We prompted GPT-4 to model 3D structures for the 20 standard amino acids and an α-helical polypeptide chain, with the latter incorporating Wolfram mathematical computation. We also used GPT-4 to perform structural interaction analysis between the anti-viral nirmatrelvir and its target, the SARS-CoV-2 main protease. Geometric parameters of the generated structures typically approximated close to experimental references. However, modeling was sporadically error-prone and molecular complexity was not well tolerated. Interaction analysis further revealed the ability of GPT-4 to identify specific amino acid residues involved in ligand binding along with corresponding bond distances. Despite current limitations, we show the current capacity of natural language generative AI to perform basic structural biology modeling and interaction analysis with atomic-scale accuracy.
Similar content being viewed by others

Regression Transformer enables concurrent sequence regression and generation for molecular language modelling
Language models can learn complex molecular distributions

Controllable protein design with language models
Introduction
Artificial intelligence (AI)-based capabilities and applications in scientific research have made remarkable progress over the past few years1,2. Advances in the field of protein structure prediction have been particularly impactful: AI-based dedicated structural biology tools such as AlphaFold2 and RoseTTAFold are capable of modeling protein structures from only amino acid sequence input with accuracy comparable to lower-resolution experimentally determined structures3,4,5. AlphaFold2 and RoseTTAFold are trained on protein sequence and structure datasets6, and rely on neural network architectures specialized for modeling protein structures. Another category of AI-based tools for protein structure prediction are protein language models, which differ from AlphaFold2 and RoseTTAFold in that they are not trained on structures but rather on protein sequences7,8,9. Collectively, such protein structure prediction tools have been extensively used by researchers across various disciplines in the biological sciences and are expected to continue to add value alongside experimental structure determination10,11,12,13,14,15,16,17,18.
On the other hand, generative AI language models, particularly the various generative pre-trained transformer (GPT) models from OpenAI19,20,<a data-track="click" data-track-action="reference anchor" data-track-label="link" data-test="citation-ref" aria-label="Reference 21" title="OpenAI. Introducing ChatGPT, (2022).” href=”https://www.nature.com/articles/s41598-024-69021-2#ref-CR21″ id=”ref-link-section-d89812522e679″>21, have garnered substantial interest in recent years. Unlike AlphaFold2, RoseTTAFold, and protein language models, GPTs are trained on natural language datasets and operate by using neural network computational architectures developed for natural language processing (NLP) rather than structural modeling. NLP involves “learning, understanding, and producing human language content” through computation22, and GPTs employ transformer-based architectures for this purpose19,20, 23. In essence, GPT architectures rely on “tokenization”, where text is broken down into smaller units referred to as tokens (ranging in size from individual letters to multiple words), and processing, which involves attention mechanisms and statistical distributions, allows for predicting the next token in a sequence of text19,20. Additional specificities about the architecture and training data are not provided in the recent GPT-4 technical report, though training data is described to consist of a large corpus of text sourced from the internet, among other natural language sources20. Interestingly, GPTs are able carry out tasks which require some level of reasoning, as demonstrated by performance in various reasoning evaluations24,25,26. Furthermore, capabilities and applications of GPTs beyond generalized NLP have been documented in various scientific disciplines, including for autonomous and predictive chemical research27,28, drug development29,30, bioinformatic analysis31,32,33, and synthetic biology34.
Our group has recently reported how GPT-4 interprets the central dogma of molecular biology and the genetic code35. While analogies can be made, the genetic code is not a natural language per se, yet GPT-4 seems to have an inherent capability of processing it. In a related line of investigation, here we explore whether GPT-4 can perform rudimentary structural biology modeling and evaluated its capabilities and limitations in this domain. Surprisingly, we found that GPT-4 is capable of modeling the 20 standard amino acids and, with incorporation of the Wolfram plugin, a typical α-helical secondary structure element at the atomic level—though not without sporadic errors. Moreover, we used GPT-4 to perform structural analysis of interaction between the anti-viral drug nirmatrelvir and its molecular target, the main protease of SARS-CoV-2 (the coronaviral etiology of COVID-19). More broadly, the findings reported here: (i) demonstrate the current capabilities of GPT-based AI in the context of structural biology modeling; (ii) highlight the performance of protein-ligand structural interaction analysis with GPT-4; and (iii) may serve as an informative reference point for comparing these capabilities as natural language-based generative AI continues to advance. To our knowledge, this is the first report to explore the structural biology modeling and interaction analysis capabilities of natural language-based generative AI.
Results
Modeling of individual amino acid structures
Amino acid residues are the components of proteins, and their atomic composition and geometric parameters have been well characterized36,37,38,39, making them suitable candidates for rudimentary structure modeling. We therefore prompted GPT-4 to model the 20 standard amino acids with minimal contextual information as input, including instructions for output in legacy Protein Data Bank (PDB) file format (Tables 1, 2, Supplementary Table S1, and Fig. 1a). GPT-3.5 was included as a performance benchmark. Multiple iterations (n = 5 for each amino acid) were run by using the same input prompt to monitor consistency (see Methods). For each individual amino acid, GPT-4 generated 3D structures with coordinate values for both backbone and sidechain atoms (Fig. 1b). Generated structures contained all atoms specific to the amino acid prompted, except for a single iteration of cysteine which lacked the backbone O atom and a single iteration of methionine which lacked the sidechain Cγ atom. Most amino acid structures (excluding achiral glycine) were modeled in L rather than D stereochemical configuration, while some were also modeled in planar configuration (Fig. 1c). While the modeling favored the L-configuration, a more accurate distribution would be near exclusive L-configuration, given that D-amino acid residues are only rarely found in naturally occurring proteins40,41.
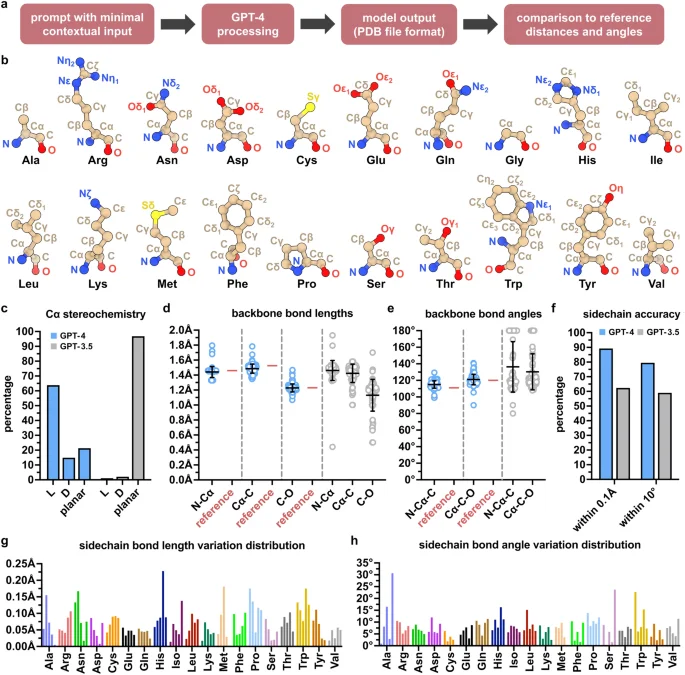
Modeling the 3D structures of the 20 standard amino acids with GPT-4. (a) Procedure for structure modeling and analysis. (b) Exemplary 3D structures of each of the 20 amino acids modeled by GPT-4. (c) Cα stereochemistry of modeled amino acids including L and D configurations as well as nonconforming planar; n = 5 per amino acid excluding achiral glycine and one GPT-4 iteration of cysteine (see Methods). (d,e) Backbone bond lengths and angles of amino acids modeled by GPT-4 (blue) relative to experimentally determined reference values (red); n = 5 per amino acid, excluding one iteration of cysteine (see Methods). Corresponding values of amino acids modeled by GPT-3.5 are shown adjacent (grey); n = 5 per amino acid. Data shown as means ± SD. (f) Sidechain accuracy of modeled amino acid structures in terms of bond lengths (within 0.1 Å) and bond angles (within 10°) relative to experimentally determined reference values; n = 5 per amino acid. See Methods for experimentally determined references. (g,h) Distributions of sidechain bond length and angle variation relative to experimentally determined reference values for each amino acid generated by GPT-4, excluding glycine. Bars represent the mean bond length or angle variation for each of the five iterations per amino acid. One of the methionine iterations was excluded (see Methods).
Backbone bond lengths and angles of the modeled structures varied in accuracy, yet clustered in approximation to experimentally determined reference values37 (Fig. 1d,e). Moreover, all reference values fell within the standard deviations of backbone bond lengths and angles of the modeled structures. Finally, sidechain bond lengths and bond angles also varied in accuracy, yet nearly 90% of calculated bond lengths were within 0.1 Å and nearly 80% of calculated bond angles were within 10° of experimentally determined reference values39,42, again indicating remarkable precision (Fig. 1f–h, Supplementary Fig. S1–S4). Sidechain bond lengths and bond angles outside of these ranges generally occurred at random, but were notably more prevalent in the aromatic rings of histidine and tryptophan, along with the pyrrolidine component of proline. Although not entirely error-free, the ring structures of phenylalanine and tyrosine were more accurate, which may be due to the reduced complexity of their all-carbon ring composition. Across all parameters assessed, GPT-4 substantially outperformed GPT-3.5. Collectively, these findings demonstrate that GPT-4 is capable of structurally modeling single amino acid residues in a manner that resembles their experimentally-determined structures, though not without sporadic errors including incorrect stereochemistry and geometric distortion, which would require—at least presently—human operator curation or supervision to ensure fidelity.
Modeling of an α-helix structure
The α-helix is the most commonly occurring and extensively studied secondary structure element found in proteins43,44,45,46. Thus, we next prompted GPT-4 and GPT-3.5 to model an α-helical polypeptide chain, but were unable to obtain accurate structures with either version, despite multiple attempts with various prompts. We then incorporated the Wolfram plugin, a mathematical computation extension developed by Wolfram-Alpha for use with GPT-447. GPT-4 used together with the Wolfram plugin was able to model a 10-residue α-helical structure and output the result in legacy PDB file format with minimal contextual information as input (Tables 1b, 2b, Supplementary Table S2, and Fig. 2a,b). Multiple iterations were run by using the same input prompt to monitor consistency, and up to two prompt-based refinements after the first attempt were permitted per iteration for improved accuracy (see Methods). To reduce complexity, only Cα atoms were modeled. Notably, prior to engaging the Wolfram plugin within the response dialog, GPT-4 often described α-helical parameters mathematically, for example:
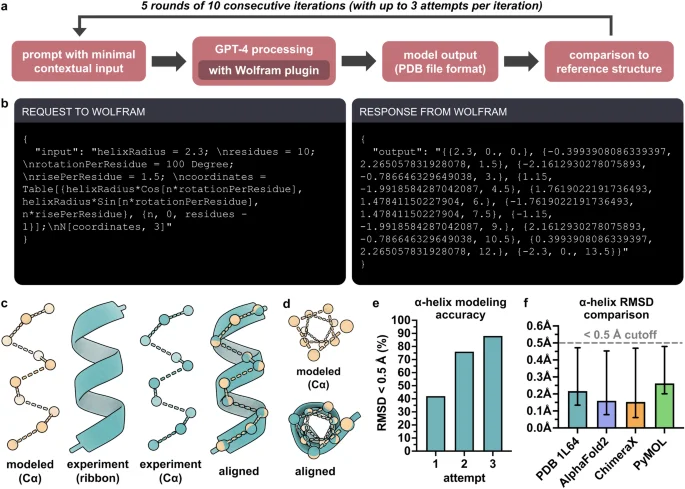
Modeling the 3D structure of an α-helical polypeptide structure with GPT-4. (a) Procedure for structure modeling and analysis. (b) Request made from GPT-4 to Wolfram and subsequent response from Wolfram to GPT-4 from an exemplary α-helix modeling iteration (also see Supplementary Table S2). (c) Exemplary 3D structure of a modeled α-helix (beige), an experimentally determined α-helix reference structure (PDB ID 1L64) (teal), and their alignment (RMSD = 0.147 Å). (d) Top-down view of modeled and experimental α-helices from panel c. (e) Accuracy of α-helix modeling as measured by number of attempts (including up to two refinements following the first attempt) required to generate a structure with RMSD < 0.5 Å relative to the experimentally determined reference structure; n = 5 rounds of 10 consecutive iterations (total n = 50 models). (f) Comparison of RMSDs between GPT-4 α-helix structures and the experimentally determined α-helix structure, the AlphaFold2 α-helix structure, the ChimeraX α-helix structure, and the PyMOL α-helix structure. Only structures with RMSD < 0.5 Å (dashed grey line) relative to each reference structure are included (88% included in reference to PDB ID 1L64; 90% to AlphaFold; 90% to ChimeraX; 88% to PyMOL). Data shown as means ± range.
“where (r) is the radius of the helix, ({theta }_{n}) is the rotation angle for the nth residue, and the rise per residue is the linear distance along the helical axis between consecutive amino acids” (Supplementary Table S2). In this case, Eqs. (1)–(3), represent the x, y, and z coordinates for the (n)th Cα atom in the α-helix and were incorporated by GPT-4 into the Wolfram request (Fig. 2b).
GPT-4 arbitrarily assigned all residues as alanine, which was likely done for the sake of simplicity, but nevertheless aligns well with the fact that alanine has the greatest α-helix propensity of all 20 standard amino acids46. Remarkably, accuracy of the modeled α-helix was comparable to an experimentally determined α-helical structure consisting of 10 consecutive alanine residues (PDB ID: 1L64)48 (Fig. 2c,d). More than 40% of modeled structures had a root-mean-square deviation (RMSD) of < 0.5 Å relative to the reference experimental structure on the first attempt, and nearly 90% had a RMSD of < 0.5 Å after two prompt-based refinements (Fig. 2e). The structures generated by GPT-4 were also compared to poly-alanine α-helix structures modeled by AlphaFold2, ChimeraX, and PyMOL, and the lowest RMSDs (i.e., greatest structural similarity) were found between the AlphaFold2 and ChimeraX structures (Fig. 2f). Taken together, these results demonstrate the capability of GPT-4, with a seamless incorporation of the Wolfram plugin, to predict the atomic level structure of an α-helix.
Structural interaction analysis
Structural interaction between drugs and proteins is a key aspect of molecular biology with basic, translational and clinical implications. For instance, binding of the Paxlovid (ritonavir-boosted nirmatrelvir) protease inhibitor compound, nirmatrelvir, to the SARS-CoV-2 main protease is of particular clinical relevance49,50, especially given the concern that the mutation-prone SARS-CoV-2 leads to treatment resistance51. Thus, we used GPT-4 to perform qualitative structural analysis of drug binding within the nirmatrelvir-SARS-CoV-2 drug-protein paradigm. We first provided the PDB file input of a crystal structure of nirmatrelvir bound to the SARS-CoV-2 main protease (PDB ID: 7VH8) 49 and prompted GPT-4 to detect the nirmatrelvir ligand, followed by a subsequent prompt for interaction detection and interaction-interfering mutation prediction (Table 1c, Supplementary Table S3, and Fig. 3a). The dialog revealed that GPT-4 engaged Python in order to perform interaction analysis, including for reading the PDB file, identifying the ligand, and parsing atomic coordinates (Supplementary Table S3).
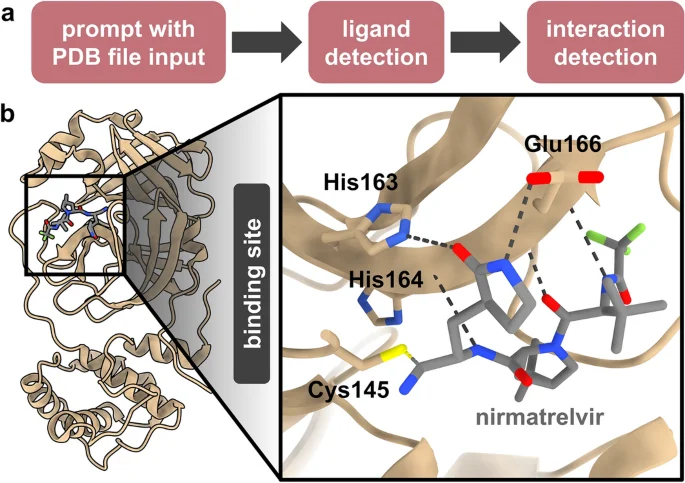
Structural analysis of interaction between nirmatrelvir and the SARS-CoV-2 main protease. (a) Procedure for performing ligand interaction analysis. (b) Crystal structure of nirmatrelvir bound to the SARS-CoV-2 main protease (PDB ID: 7VH8) with bond-forming residues detected by GPT-4, and their bonds depicted with ChimeraX (inset). Distances between interacting atom pairs were 1.81 Å (Cys145 Sγ–C3), 2.68 Å (His163 Nε2–O1), 2.77 Å (Glu166 O–N4), 3.02 Å (His164 O–N1), as determined by GPT-4 and 1.814 Å (Cys145 Sγ–C3), 2.676 Å (His163 Nε2–O1), 2.767 Å (Glu166 O–N4), 2.851 Å (Glu166 N–O3), 3.019 Å (Glu166 Oε1–N2), 3.017 Å (His164 O–N1), as determined with ChimeraX. Note that distance values corresponding to the Glu166 N–O3 and Glu166 Oε1–N2 atom pair interactions were not provided by GPT-4.
GPT-4 correctly identified the nirmatrelvir ligand, which in the input PDB file is designated as “4WI” (Supplementary Table S3). For interaction detection, GPT-4 listed five amino acid residues within the substrate-binding pocket of the protein, four of which directly bind the nirmatrelvir ligand (Cys145 forms a covalent bond, His163 and His164 each form hydrogen bonds, and Glu166 forms three separate hydrogen bonds)49 (Fig. 3b). The fifth residue (Thr190) does not form a bond with the ligand, but is located within the binding pocket49. Moreover, the distances provided by GPT-4 for the four binding residues correspond precisely to the distances between the interacting atoms, information which is not inherent in the input PDB file. GPT-4 also described several mutations which may interfere with binding (Supplementary Table S3), and while most were plausible, others would likely be inconsequential. Notably, however, the suggested mutation of Glu166 to a residue lacking negative charge has been documented to be critically detrimental to nirmatrelvir binding52,53,54 and confers clinical therapeutic resistance55,56. Altogether, this exercise reveals the ability of GPT-4 to perform basic structural analysis of protein-ligand interaction in a manner which, in conjunction with molecular analysis software such as ChimeraX, highlights its potential for practical utility.
Discussion
The exploratory findings reported here demonstrate the current capabilities and limitations of GPT-4, a natural language-based generative AI, for rudimentary structural biology modeling and drug interaction analysis. This presents a unique aspect of novelty, given the inherent distinction between natural language models and other dedicated AI tools commonly used for structural biology, including AlphaFold, RoseTTAFold, and protein language models. While such tools are unequivocally far more sophisticated in terms of the scale of molecular complexity that they are able to process, GPT-4 sets the stage for a broadly accessible and computationally distinct avenue for use in structural biology. However, there are substantial improvements needed before the GPT family of language models may reliably provide advanced practical utility in this domain. The current rudimentary modeling capabilities, while notable, must evolve such that modeling of higher complexity biomolecular structures, including unique structural motifs and tertiary structure, could be performed. Meanwhile, the rudimentary capabilities documented here provide precedent for more complex modeling, and may serve to inform future evaluations amidst ongoing advancements in natural language-based generative AI technology.
The performance of GPT-4 for modeling of the 20 standard amino acids was favorable in terms of atom composition, bond lengths, and bond angles. However, stereochemical configuration propensity and modeling of ring structures require improvement. Performance for α-helix modeling, with a seamless incorporation of advanced mathematical computation from the Wolfram plugin, was also favorable. While the requirement of prompt-based refinements may be viewed as a limitation, they may also serve as a means and opportunity by which the user can optimize and modify a structure. Nonetheless, improvements will be required in the capacity to model more complex all-atom structures, not only Cα backbone atoms. Moreover, the sporadic occurrence of errors should not be taken lightly, as introduction of errors at even the smallest scale may be highly detrimental to any structural model and associated biological interpretations.
These structural modeling capabilities also raise the question of modeling methodology, especially since GPT-4 was not explicitly developed for this specialized purpose. It would be challenging to provide a precise answer for this, and several computational methods may be involved. For instance, GPT-4 may be utilizing pre-existing atomic coordinate information present in its broad training dataset, which includes “publicly available data (such as internet data) and data licensed from third-party providers”20. However, this reasoning does not adequately explain the geometric variability observed in the predicted structures, and why structural complexity appears to be a limiting factor. The modeling may also be performed ab initio, given that the generated responses often articulate geometric parameters (e.g., specific bond lengths and angles, number of amino acid residues per α-helix turn, α-helix diameter, etc.) in addition to providing atomic coordinates (Supplementary Tables S1, S2). Alternatively, the modeling methodology may involve both the use of pre-existing coordinates plus ab initio computation.
Of note, the comparison of the α-helix model generated by GPT-4 with those generated by other computational tools was quite revealing. AlphaFold2, as mentioned above, predicts structures based on training data consisting of protein sequences and 3D structures, and was developed specifically for modeling protein structures. In addition to their dedicated molecular analysis capabilities, ChimeraX and PyMOL may be used to model basic, idealized secondary structure elements in a manner which narrowly considers precise predefined geometries, thus providing accurate α-helix structures. Despite not being explicitly developed to model atomic coordinates for α-helical segments of protein chains, GPT-4 was able to generate an α-helix with accuracy comparable to the structures modeled by the above tools. The requirement of adding the Wolfram plugin likely suggests that mathematical computation is heavily relied upon by GPT-4 for α-helix modeling. Yet, α-helix structural properties and self-instruction are generated by GPT-4 prior to engaging the Wolfram plugin (Supplementary Table S2), suggesting that some degree of intrinsic “reasoning” might perhaps be involved. So-called reasoning, in this regard, is in reference to the documented performance of GPTs in various reasoning evaluations19,20, 24,25,26, and it should be noted that there is ongoing debate about what constitutes reasoning as it pertains to AI57,58.
The exercise exploring the capability of GPT-4 to perform structural analysis of ligand-protein binding showed promise, especially given the clinical relevance of the protein binding interaction between nirmatrelvir and the SARS-CoV-2 main protease. Ligand detection was expected to be a straightforward task, as PDB files include unique designations for various molecular entities. Interaction detection was surprisingly well-handled, considering the complexity of locating amino acid residues with spatial proximity to the ligand and providing precise distances between interacting atoms. Based on the generated response (Supplementary Table S3), it is likely that proximity was the primary criterion used by GPT-4 for interaction detection. While proximity is important, the analysis would benefit from additional criteria such as hydrophobicity, electrostatic potential, solvent effects, etc. Moreover, if the analytical capabilities of GPTs improve such that multiple interaction criteria are considered simultaneously and automatically (i.e., without specific user instruction), far more comprehensive structural interaction analysis would likely be achievable. Finally, the prediction of interaction-interfering mutations may become particularly useful in drug discovery and development, an area where GPT-based AI is anticipated to be impactful59,60,61.
Considering both strengths and weaknesses, the structural modeling capabilities of GPT represent an intriguing aspect of the unprecedented advancement of natural language-based generative AI, a transformative technology presumably still in its infancy. While this modeling remains rudimentary and is currently of limited practical utility, it establishes an immediate and direct precedent for applying this technology in structural biology as generative AI natural language models undergo continued development and specialization. Concurrently, this broadly-accessible technology presents opportunity for structural analysis of drug-protein interaction. In the interim, further research on the capabilities and limitations of generative AI is merited, not only in structural biology but also for other potential applications in the biological sciences.
Methods
Prompt-based modeling with GPT-4
Modeling of individual amino acid structures was performed by challenging GPT-4 through the ChatGPT interface20,<a data-track="click" data-track-action="reference anchor" data-track-label="link" data-test="citation-ref" aria-label="Reference 21" title="OpenAI. Introducing ChatGPT, (2022).” href=”https://www.nature.com/articles/s41598-024-69021-2#ref-CR21″ id=”ref-link-section-d89812522e1688″>21 with a single prompt (Table 1a), one amino acid residue at a time. For each individual amino acid, the same prompt was used for five consecutive iterations with each iteration initiated in a new dialog. GPT-4 was run in classic mode without “browser” and “analysis” features enabled, formerly known as “web browser” and “code interpreter” plug-ins, respectively61. Classic mode limits processing to GPT-4 with no additional capabilities. Amino acid modeling was also performed with GPT-3.5 in the same manner. However, GPT-3.5 would frequently generate PDB file output with missing or extra atoms. In such cases, responses were regenerated within each GPT-3.5 dialog until PDB file output contained the correct number of atoms required for analysis. Modeling of α-helix structures was performed by challenging GPT-4 running the Wolfram plugin47 through the ChatGPT interface with an initial prompt followed by up to two refinement prompts in the same dialog, for a total of up to three attempts (Table 1b). The same prompt was used for five rounds of ten consecutive iterations with each iteration initiated in a new dialog.
Analysis of generated structures
Structures were analyzed by using UCSF ChimeraX42. For amino acid structures, the “distance” and “angle” commands were used for determining bond lengths and bond angles, respectively. These commands were tailored for each amino acid type in order to account for sidechain atom specificity (Supplementary Table S5). Experimentally determined reference values for backbone bond lengths (N-Cα, 1.459 Å; Cα-C, 1.525 Å; C-O 1.229 Å) and backbone bond angles (N-Cα-C, 111.0°; Cα-C-O, 120.1°), as depicted in Fig. 1d,e, were previously established by protein structure X-ray diffraction statistical analyses37. While backbone geometry is conformationally dependent, idealized reference values were used in the current study for simplicity38. Experimentally determined sidechain bond lengths and angles (Supplementary Table S4) were obtained from a backbone-dependent rotamer library built into ChimeraX, with dihedral angles set to φ = 180°, ψ = 180°, and ω = 180° (representative of a fully extended backbone in trans configuration)39,42. For GPT-4 amino acid modeling, one iteration of cysteine lacked the backbone O atom and one iteration of methionine lacked the sidechain Cγ atom. Thus, these single iterations (n = 1) were excluded from analyses involving the missing atoms.
For α-helix structures, the matchmaker tool within ChimeraX was used for alignment and RMSD determination. The matchmaker tool was run with default parameters for chain pairing (i.e., best-aligning pair of chains between reference and match structure), alignment (i.e., Needleman-Wunsch sequence alignment algorithm and BLOSUM-62 matrix), and fitting (i.e., iteration by pruning long atom pairs with an iteration cutoff distance of 2.0 Å). An α-helical structure consisting of 10 consecutive alanine residues, detected within an engineered form of bacteriophage T4 lysozyme resolved by X-ray diffraction (PDB ID 1L64)48, was used as the experimental reference for evaluating the α-helix structures modeled by GPT-4. The AlphaFold2 α-helix structure was modeled using ColabFold62 through ChimeraX by using the built-in AlphaFold interface. An elongated polyalanine sequence was used in order to meet the minimum input requirements and prediction was run with default parameters (i.e., without PDB template use and without energy minimization) (Supplementary Table S6 and Supplementary Fig. S5a). The two ChimeraX and PyMOL α-helix structures, were modeled by using the build structure command (within ChimeraX) and fab command (within PyMOL63), respectively, each by using a 10-residue alanine sequence as input and run with default α-helix parameters (i.e., backbone dihedral angles set to φ = −57° and ψ = −47°) (Supplementary Fig. S5b,c). The AlphaFold2, ChimeraX, and PyMOL α-helix structures were all exported in PDB file format for comparison with GPT-4 structures. All data were analyzed by using GraphPad Prism 10.1.0 (GraphPad Software). Statistical details are reported in the figure legends and statistical measurements include mean, mean ± SD, and mean ± range.
Prompt-based interaction analysis with GPT-4
Structural analysis of binding interaction was performed by providing GPT-4 with an input PDB file and prompting as described (Table 1B) through the ChatGPT interface. The PDB file used as input was unmodified as obtained from the PDB entry for PDB ID: 7VH849. It should be noted that PDB ID: 7VH8 refers to nirmatrelvir as PF-07321332. For this exercise, GPT-4 was not limited to classic mode. Rather the “browser” and “analysis” features were enabled within the ChatGPT interface to enable file input, a feature available for GPT-4 but not GPT-3.5. Only the “analysis” feature was engaged for the responses generated by GPT-4 (Supplementary Table S3). ChimeraX was used to analyze amino acid residues detected by GPT-4 to interact with nirmatrelvir. The “contacts” tool was run with the five specific residues identified by GPT-4 (Supplementary Table S3) and the nirmatrelvir molecule under selection within the 7VH8 PDB structure. The “contacts” command was run with default parameters (i.e., van der Waals (VDW) overlap ≥ −0.4 Å) limited to the selected residues and nirmatrelvir in order to identify interacting atom pairs between them as well as corresponding distance values.
Data availability
All data are available from the corresponding authors upon request.
References
-
Jordan, M. I. & Mitchell, T. M. Machine learning: Trends, perspectives, and prospects. Science 349, 255–260. https://doi.org/10.1126/science.aaa8415 (2015).
Google Scholar
-
Wang, H. et al. Scientific discovery in the age of artificial intelligence. Nature 620, 47–60. https://doi.org/10.1038/s41586-023-06221-2 (2023).
Google Scholar
-
Jumper, J. et al. Highly accurate protein structure prediction with alphafold. Nature 596, 583–589. https://doi.org/10.1038/s41586-021-03819-2 (2021).
Google Scholar
-
Baek, M. et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876. https://doi.org/10.1126/science.abj8754 (2021).
Google Scholar
-
Kryshtafovych, A., Schwede, T., Topf, M., Fidelis, K. & Moult, J. Critical assessment of methods of protein structure prediction (CASP)-round XIV. Proteins 89, 1607–1617. https://doi.org/10.1002/prot.26237 (2021).
Google Scholar
-
Burley, S. K. & Berman, H. M. Open-access data: A cornerstone for artificial intelligence approaches to protein structure prediction. Structure 29, 515–520. https://doi.org/10.1016/j.str.2021.04.010 (2021).
Google Scholar
-
Elnaggar, A. et al. ProtTrans: Toward understanding the language of life through self-supervised learning. IEEE Trans. Pattern Anal. Mach. Intell. 44, 7112–7127. https://doi.org/10.1109/TPAMI.2021.3095381 (2022).
Google Scholar
-
Chowdhury, R. et al. Single-sequence protein structure prediction using a language model and deep learning. Nat. Biotechnol. 40, 1617–1623. https://doi.org/10.1038/s41587-022-01432-w (2022).
Google Scholar
-
Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130. https://doi.org/10.1126/science.ade2574 (2023).
Google Scholar
-
Bordin, N. et al. AlphaFold2 reveals commonalities and novelties in protein structure space for 21 model organisms. Commun. Biol. 6, 160. https://doi.org/10.1038/s42003-023-04488-9 (2023).
Google Scholar
-
Bordin, N. et al. Novel machine learning approaches revolutionize protein knowledge. Trends Biochem. Sci. 48, 345–359. https://doi.org/10.1016/j.tibs.2022.11.001 (2023).
Google Scholar
-
Mosalaganti, S. et al. AI-based structure prediction empowers integrative structural analysis of human nuclear pores. Science 376, eabm9506. https://doi.org/10.1126/science.abm9506 (2022).
Google Scholar
-
Fontana, P. et al. Structure of cytoplasmic ring of nuclear pore complex by integrative cryo-EM and alphafold. Science 376, eabm9326. https://doi.org/10.1126/science.abm9326 (2022).
Google Scholar
-
Read, R. J., Baker, E. N., Bond, C. S., Garman, E. F. & van Raaij, M. J. AlphaFold and the future of structural biology. Acta Crystallogr. F Struct. Biol. Commun. 79, 166–168. https://doi.org/10.1107/S2053230X23004934 (2023).
Google Scholar
-
Edich, M., Briggs, D. C., Kippes, O., Gao, Y. & Thorn, A. The impact of AlphaFold2 on experimental structure solution. Faraday Discuss. 240, 184–195. https://doi.org/10.1039/d2fd00072e (2022).
Google Scholar
-
Varadi, M. et al. AlphaFold protein structure database in 2024: Providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. https://doi.org/10.1093/nar/gkad1011 (2023).
Google Scholar
-
Varadi, M. & Velankar, S. The impact of AlphaFold protein structure database on the fields of life sciences. Proteomics 23, e2200128. https://doi.org/10.1002/pmic.202200128 (2023).
Google Scholar
-
Burley, S. K., Arap, W. & Pasqualini, R. Predicting proteome-scale protein structure with artificial intelligence. N. Engl. J. Med. 385, 2191–2194. https://doi.org/10.1056/NEJMcibr2113027 (2021).
Google Scholar
-
Brown, T. B. et al. Language models are few-shot learners. arXiv https://doi.org/10.48550/arXiv.2005.14165 (2020).
-
OpenAI. GPT-4 Technical Report. arXiv https://doi.org/10.48550/arXiv.2303.08774 (2023).
-
OpenAI. Introducing ChatGPT, <https://openai.com/blog/chatgpt> (2022).
-
Hirschberg, J. & Manning, C. D. Advances in natural language processing. Science 349, 261–266. https://doi.org/10.1126/science.aaa8685 (2015).
Google Scholar
-
Vaswani, A. et al. Attention is all you need. arXiv https://doi.org/10.48550/arXiv.1706.03762 (2017).
-
Webb, T., Holyoak, K. J. & Lu, H. Emergent analogical reasoning in large language models. Nat. Hum. Behav. 7, 1526–1541. https://doi.org/10.1038/s41562-023-01659-w (2023).
Google Scholar
-
Hagendorff, T., Fabi, S. & Kosinski, M. Human-like intuitive behavior and reasoning biases emerged in large language models but disappeared in ChatGPT. Nat. Comput. Sci. 3, 833–838. https://doi.org/10.1038/s43588-023-00527-x (2023).
Google Scholar
-
Yax, N., Anlló, H. & Palminteri, S. Studying and improving reasoning in humans and machines. Commun. Psychol. 2, 51. https://doi.org/10.1038/s44271-024-00091-8 (2024).
-
Boiko, D. A., MacKnight, R., Kline, B. & Gomes, G. Autonomous chemical research with large language models. Nature 624, 570–578. https://doi.org/10.1038/s41586-023-06792-0 (2023).
Google Scholar
-
Jablonka, K. M., Schwaller, P., Ortega-Guerrero, A. & Smit, B. Leveraging large language models for predictive chemistry. Nat. Mach. Intell. https://doi.org/10.1038/s42256-023-00788-1 (2024).
-
Savage, N. Drug discovery companies are customizing ChatGPT: Here’s how. Nat. Biotechnol. 41, 585–586. https://doi.org/10.1038/s41587-023-01788-7 (2023).
Google Scholar
-
Wang, R., Feng, H. & Wei, G. W. ChatGPT in drug discovery: A case study on anticocaine addiction drug development with chatbots. J. Chem. Inform. Model. 63, 7189–7209. https://doi.org/10.1021/acs.jcim.3c01429 (2023).
Google Scholar
-
Lubiana, T. et al. Ten quick tips for harnessing the power of ChatGPT in computational biology. PLoS Comput. Biol. 19, e1011319. https://doi.org/10.1371/journal.pcbi.1011319 (2023).
Google Scholar
-
Shue, E. et al. Empowering beginners in bioinformatics with ChatGPT. Quant. Biol. 11, 105–108. https://doi.org/10.15302/j-qb-023-0327 (2023).
Google Scholar
-
Karkera, N., Acharya, S. & Palaniappan, S. K. Leveraging pre-trained language models for mining microbiome-disease relationships. BMC Bioinform. 24, 290. https://doi.org/10.1186/s12859-023-05411-z (2023).
-
Xiao, Z. et al. Generative artificial intelligence GPT-4 accelerates knowledge mining and machine learning for synthetic biology. ACS Synth. Biol. 12, 2973–2982. https://doi.org/10.1021/acssynbio.3c00310 (2023).
Google Scholar
-
Ille, A. M. & Mathews, M. B. AI interprets the central dogma and genetic code. Trends Biochem. Sci. 48, 1014–1018. https://doi.org/10.1016/j.tibs.2023.09.004 (2023).
Google Scholar
-
Engh, R. A. & Huber, R. Accurate bond and angle parameters for X-ray protein structure refinement. Acta Crystallogr. Sect. A 47, 392–400. https://doi.org/10.1107/S0108767391001071 (1991).
Google Scholar
-
Engh, R. A. & Huber, R. International Tables for Crystallography Volume F: Crystallography of Biological Macromolecules (Springer, 2001).
-
Berkholz, D. S., Shapovalov, M. V., Dunbrack, R. L. Jr. & Karplus, P. A. Conformation dependence of backbone geometry in proteins. Structure 17, 1316–1325. https://doi.org/10.1016/j.str.2009.08.012 (2009).
Google Scholar
-
Shapovalov, M. V. & Dunbrack, R. L. Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 19, 844–858. https://doi.org/10.1016/j.str.2011.03.019 (2011).
Google Scholar
-
Fujii, N. & Saito, T. Homochirality and life. Chem. Rec. 4, 267–278. https://doi.org/10.1002/tcr.20020 (2004).
Google Scholar
-
Mitchell J. B. O. & Smith, J. D‐amino acid residues in peptides and proteins. Proteins 50, 563–571. https://doi.org/10.1002/prot.10320 (2003).
-
Meng, E. C. et al. UCSF chimeraX: Tools for structure building and analysis. Protein Sci. 32, e4792. https://doi.org/10.1002/pro.4792 (2023).
Google Scholar
-
Doig, A. J. et al. Structure, stability and folding of the alpha-helix. Biochem. Soc. Symp. https://doi.org/10.1042/bss0680095 (2001).
Google Scholar
-
Eisenberg, D. The discovery of the alpha-helix and beta-sheet, the principal structural features of proteins. Proc. Natl. Acad. Sci. USA 100, 11207–11210. https://doi.org/10.1073/pnas.2034522100 (2003).
Google Scholar
-
Pauling, L., Corey, R. B. & Branson, H. R. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA 37, 205–211. https://doi.org/10.1073/pnas.37.4.205 (1951).
Google Scholar
-
Pace, C. N. & Scholtz, J. M. A helix propensity scale based on experimental studies of peptides and proteins. Biophys. J. 75, 422–427. https://doi.org/10.1016/s0006-3495(98)77529-0 (1998).
Google Scholar
-
Wolfram, S. ChatGPT Gets Its “Wolfram Superpowers”! https://writings.stephenwolfram.com/2023/03/chatgpt-gets-its-wolfram-superpowers/ (2023).
-
Heinz, D. W., Baase, W. A. & Matthews, B. W. Folding and function of a T4 lysozyme containing 10 consecutive alanines illustrate the redundancy of information in an amino acid sequence. Proc. Natl. Acad. Sci. USA 89, 3751–3755. https://doi.org/10.1073/pnas.89.9.3751 (1992).
Google Scholar
-
Zhao, Y. et al. Crystal structure of SARS-CoV-2 main protease in complex with protease inhibitor PF-07321332. Protein Cell 13, 689–693. https://doi.org/10.1007/s13238-021-00883-2 (2022).
Google Scholar
-
Hammond, J. et al. Oral nirmatrelvir for high-risk, nonhospitalized adults with Covid-19. N. Engl. J. Med. 386, 1397–1408. https://doi.org/10.1056/NEJMoa2118542 (2022).
Google Scholar
-
Chatterjee, S., Bhattacharya, M., Dhama, K., Lee, S. S. & Chakraborty, C. Resistance to nirmatrelvir due to mutations in the Mpro in the subvariants of SARS-CoV-2 omicron: Another concern?. Mol. Ther. Nucleic Acids 32, 263–266. https://doi.org/10.1016/j.omtn.2023.03.013 (2023).
Google Scholar
-
Hu, Y. et al. Naturally occurring mutations of SARS-CoV-2 main protease confer drug resistance to nirmatrelvir. ACS Cent. Sci. 9, 1658–1669. https://doi.org/10.1021/acscentsci.3c00538 (2023).
Google Scholar
-
Iketani, S. et al. Multiple pathways for SARS-CoV-2 resistance to nirmatrelvir. Nature 613, 558–564. https://doi.org/10.1038/s41586-022-05514-2 (2023).
Google Scholar
-
Zhou, Y. et al. Nirmatrelvir-resistant SARS-CoV-2 variants with high fitness in an infectious cell culture system. Sci. Adv. 8, eadd7197. https://doi.org/10.1126/sciadv.add7197 (2022).
Google Scholar
-
Zuckerman, N. S., Bucris, E., Keidar-Friedman, D., Amsalem, M. & Brosh-Nissimov, T. Nirmatrelvir resistance-de novo E166V/L50V mutations in an immunocompromised patient treated with prolonged nirmatrelvir/ritonavir monotherapy leading to clinical and virological treatment failure-a case report. Clin. Infect. Dis. https://doi.org/10.1093/cid/ciad494 (2023).
-
Hirotsu, Y. et al. Multidrug-resistant mutations to antiviral and antibody therapy in an immunocompromised patient infected with SARS-CoV-2. Med 4(813–824), e814. https://doi.org/10.1016/j.medj.2023.08.001 (2023).
Google Scholar
-
Eisenstein, M. A test of artificial intelligence. Nature https://doi.org/10.1038/d41586-023-02822-z (2023).
Google Scholar
-
Biever, C. ChatGPT broke the turing test—The race is on for new ways to assess AI. Nature 619, 686–689. https://doi.org/10.1038/d41586-023-02361-7 (2023).
Google Scholar
-
Chakraborty, C., Bhattacharya, M. & Lee, S. S. Artificial intelligence enabled ChatGPT and large language models in drug target discovery, drug discovery, and development. Mol. Ther. Nucleic Acids 33, 866–868. https://doi.org/10.1016/j.omtn.2023.08.009 (2023).
Google Scholar
-
Gurwitz, D. & Shomron, N. Artificial intelligence utility for drug development: ChatGPT and beyond. Drug Dev. Res. https://doi.org/10.1002/ddr.22121 (2023).
Google Scholar
-
OpenAI. ChatGPT plugins. https://openai.com/blog/chatgpt-plugins#code-interpreter (2023).
-
Mirdita, M. et al. ColabFold: Making protein folding accessible to all. Nat. Methods 19, 679–682. https://doi.org/10.1038/s41592-022-01488-1 (2022).
Google Scholar
-
Schrodinger, LLC. The PyMOL Molecular Graphics System, Version 2.5.7. https://www.pymol.org/ (2023).
Acknowledgements
This work was supported by core services from the Cancer Center Support Grant of the Rutgers Cancer Institute (P30CA072720), by the National Institutes of Health (R01CA226537 to R.P. and W.A.), and by the Levy-Longenbaugh Donor-Advised Fund (to R.P. and W.A.). RCSB Protein Data Bank is jointly funded by the National Science Foundation (DBI-1832184, PI: S.K.B.), the US Department of Energy (DE-SC0019749, PI: S.K.B.), and the National Cancer Institute, the National Institute of Allergy and Infectious Diseases, and the National Institute of General Medical Sciences of the National Institutes of Health (R01GM133198, PI: S.K.B.). Molecular graphics and analyses performed with UCSF ChimeraX, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases.
Author information
Authors and Affiliations
Contributions
A.M.I. conceptualization; A.M.I., C.M., and S.K.B. methodology; A.M.I. and C.M. investigation; A.M.I. data curation; A.M.I., C.M., S.K.B., M.B.M., R.P., and W.A. formal analysis; A.M.I. and C.M. writing—original draft; S.K.B., M.B.M., R.P., and W.A. writing—review & editing; S.K.B., R.P., and W.A. funding acquisition; R.P. and W.A. overall project supervision.
Corresponding authors
Ethics declarations
Competing interests
A.M.I. is a founder and partner of North Horizon, which is engaged in the development of artificial intelligence-based software. C.M. declares no competing interests. S.K.B. declares no competing interests. M.B.M. declares no competing interests. R.P. is a founder and equity shareholder of PhageNova Bio. R.P. is Chief Scientific Officer and a paid consultant of PhageNova Bio. R.P. is a founder and equity shareholder of MBrace Therapeutics. R.P. serves as a paid consultant for MBrace Therapeutics. R.P. has Sponsored Research Agreements (SRAs) in place with PhageNova Bio and with MBrace Therapeutics. These arrangements are managed in accordance with the established institutional conflict-of-interest policies of Rutgers, The State University of New Jersey. This study falls outside of the scope of these SRAs. W.A. is a founder and equity shareholder of PhageNova Bio. W.A. is a founder and equity shareholder of MBrace Therapeutics. W.A. serves as a paid consultant for MBrace Therapeutics. W.A. has Sponsored Research Agreements (SRAs) in place with PhageNova Bio and with MBrace Therapeutics. These arrangements are managed in accordance with the established institutional conflict-of-interest policies of Rutgers, The State University of New Jersey. This study falls outside of the scope of these SRAs.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Supplementary Information.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
Reprints and permissions
About this article
Cite this article
Ille, A.M., Markosian, C., Burley, S.K. et al. Generative artificial intelligence performs rudimentary structural biology modeling.
Sci Rep 14, 19372 (2024). https://doi.org/10.1038/s41598-024-69021-2
-
Received: 26 March 2024
-
Accepted: 30 July 2024
-
Published: 21 August 2024
-
DOI: https://doi.org/10.1038/s41598-024-69021-2
Keywords
- Artificial intelligence
- GPT
- Language model
- Machine learning
- Protein modeling
- Structural biology
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.