Abstract
Background
Although polygenic risk scores (PRSs) are expected to be helpful in precision medicine, it remains unclear whether high-PRS groups are more likely to benefit from preventive interventions for diseases. Recent methodological advancements enable us to predict treatment effects at the individual level.
Methods
We employed causal forest to explore the relationship between PRSs and individual risk of diseases associated with certain environmental factors. Following simulations illustrating its performance, we applied our approach to investigate the individual risk of cardiometabolic diseases, including coronary artery diseases (CAD) and type 2 diabetes (T2D), associated with obesity and smoking among individuals from UK Biobank (UKB; n = 369,942) and BioBank Japan (BBJ; n = 149,421).
Results
Here we find the heterogeneous association of obesity and smoking with diseases across PRS values, complicated by the multi-dimensional combination of individual characteristics such as age and sex. The highest positive correlations of PRSs and the exposure-related disease risks are observed between obesity and T2D in UKB and between smoking and CAD in BBJ (Spearman’s ρ = 0.61 and 0.32, respectively). However, most relationships are weak or negative, suggesting that high-PRS groups will not necessarily benefit most from environmental factor prevention.
Conclusions
Our study highlights the importance of individual-level prediction of disease risks associated with target exposure in precision medicine.
Plain language summary
This study aimed to understand if people with a high genetic risk for certain diseases benefit more from preventive strategies. Using a machine-learning-based method, we analyzed data from large groups of people in the UK and Japan. We examined the risk of heart and metabolic diseases in relation to obesity and smoking. The results showed that the link between genetic risk and disease is complex and varies widely among individuals. Our results suggested that those with a high genetic risk for disease may not always benefit more from the prevention of obesity and smoking. This finding suggests that we need to consider more than risk in decisions on how to prevent diseases in individuals.
Introduction
Chronic diseases impose an enormous health and economic burden. For instance, cardiovascular diseases and diabetes cause 17.9 million and 1.5 million deaths worldwide in 2019, respectively1,2. As an essential part of the Sustainable Development Goals by 20303, the United Nations has proposed to reduce premature mortality from noncommunicable diseases including cardiovascular disease and diabetes by one-third. To achieve this goal, it is imperative to establish an individualized approach to effectively reduce the risk of these diseases through improving health behaviors (e.g., physical activity, smoking, etc.)4,5. Over the last decade, genome-wide association studies (GWAS) have uncovered the contribution of genetic variants to the development of these diseases with lifestyle/behavioral risk factors6,7. Recent progress in GWAS has enabled us to summarize the individual genetic variants into a single liability score to develop a certain disease as polygenic risk scores (PRSs). PRSs can predict a high-risk group for the disease and are expected to be helpful to improve the quality of precision medicine8.
However, we still lack insight into how to maximize the advantage of PRSs for precision preventive medicine to motivate behavioral changes9. Should we simply concentrate behavioral interventions on individuals with high PRS values? To answer this question, it would be essential to obtain knowledge on how PRSs are associated with the effects of health behavior-related environmental risk factors on diseases. Previous studies applied approaches that regress interaction terms between environmental risk factors and PRSs linearly to the disease risks or assess the associations between environmental risk factors and PRSs stratified by the values of PRSs10,11,12,13. While such parametric or stratified analyses are informative when there is prior knowledge on the structures of associations, they are likely to miss multi-dimensional, complex, and non-linear heterogeneous patterns in the association that the investigators do not know in advance. Moreover, assessing the continuous relationship between PRSs and benefit of behavioral changes (i.e., without setting specific cut-off of PRSs a priori for stratification) is challenging to such conventional approaches. Given that shared decision-making between clinicians and patients requires information on absolute effect measures (e.g., risk difference)14, it is imperative to investigate whether the risk difference differs across individuals taking account of a high-dimensional set of covariates including continuous PRSs.
Recent methodological advancement in the machine-learning-based approach enables us to estimate heterogeneous treatment effects (HTEs)15. HTEs refer to situations when the effects of the exposure on the outcome (or the exposure–outcome associations) at the individual levels, known as individualized treatment effects (ITEs), vary by individual characteristics. Causal forest is one such method using random forest with double-sample trees to identify the heterogeneity in the treatment effect on an absolute scale (i.e., risk difference)16,17. In addition, this non-parametric approach allows us to predict the ITEs (in randomized clinical trials16,17) or the individual disease risk associated with exposure (in observational studies18,19) as a function of observable characteristics of individuals.
Here, we propose an approach to utilize the machine-learning causal forest model to gain insight into whether high-PRS groups are more likely to benefit from preventive interventions. The machine-learning-based approach is advantageous in that it can disentangle continuous and non-linear relationships between the PRS of a disease and the treatment effect of an environmental factor, complicated by other characteristics. This study comprises two main components: (1) simulations and (2) the application of our approach to real-world biobank data. In the simulations, we illustrate the conceptual framework of our study and demonstrate the methodological advancement of the machine-learning-based approach using simulated data. In the application to biobanks, we investigate the continuous relationship between PRSs and the estimated risk of coronary artery diseases (CAD) and its risk factors—type 2 diabetes (T2D), dyslipidemia (DL), and hypertension (HTN)—associated with obesity and smoking (two major environmental risk factors)20 using the causal forest model. We target individuals with two different ancestries from two large-scale nation-wide biobanks: UK Biobank (UKB) and BioBank Japan (BBJ). Revealing these relationships would provide a novel insight into the utility of PRSs for precision medicine to effectively prevent diseases with lifestyle/behavioral risk factors.
Methods
Estimation of heterogeneity in the association between environmental risk factors and diseases using causal forest
For simulation and real-world biobank data, we applied the causal forest model (grf package in R) to build the models to predict the ITE of environmental risk factors17. Formally, within the counterfactual framework, the effect of environmental risk factors on diseases conditional on a set of covariates (C = c) can be written as follows;
where Yx denotes the potential outcome Y under the treatment (exposure) X = x. To obtain unbiased estimates, we need the assumptions of conditional exchangeability (i.e., Yx ⫫ X | C = c), positivity (i.e., P(X = x | C = c) > 0 for all x and c), consistency (i.e., Yx = Y when X = x), no model misspecification, and no other sources of bias (e.g., misclassification, no interference, etc.).
In the causal forest approach, we constructed an ensemble of 2000 causal trees that identified subgroups with different magnitudes of the associations by individual characteristics or covariates. In each leaf of the trees, the covariate balance between treatment (exposed) and control (unexposed) groups was enssured under the assumption of no unmeasured confounders. To minimize the risk of overfitting, we employed the following two steps of the double-sample trees approach when building each tree using observable individual characteristics17: (i) randomly select the half subsample without replacement from the entire dataset to build each tree, and (ii) further split the fractional subsample into halves and used the first half to construct the tree and the second half to make predictions, so-called honest estimation16. The models were built by tenfold cross-fitting, and therefore, estimates for each fold were calculated based on trees that were fit without observations from that fold21. The calibration performance of the causal forest models was evaluated through computing the best linear fit of the target estimand using the out-of-bag prediction and the mean forest prediction as the sole two regressors18. In the best linear fit model, the forest was considered to capture heterogeneity when the coefficient of the out-of-bag prediction (termed as calibration coefficient) was significantly greater than 0 and close to 1. Further details on the causal forest approach can be found elsewhere16,17.
We evaluated the correlation between PRS and ITEs or individual risks using Spearman’s correlation coefficient. The positive correlations mentioned in the main text were significant even after multiple testing correction in the biobank analyses, unless otherwise specified.
Simulation
Data generation
We conducted Monte Carlo simulation for the varying relationships between PRSs and treatment effects of environmental factors on traits to illustrate the conceptual backgrounds of this study and methodological advantages of causal forest. We simulated n individuals with a binary outcome trait (Y) generated from a logistic regression model of an additive effect of PRS (PRS), a binary environmental factor (E), and covariates (Xi) with coefficients and an interaction effect according to different situations as
where I and CY are an interaction term and intercept, respectively. CY was determined based on the prevalence of trait, pY. PRS values were generated from the standard normal distribution22. We simulated three different situations: (1) a simple model (i.e., no interaction effect); (2) a model is complicated by an interaction effect between PRS and an environmental factor on the outcome trait: i.e., I = log(aint) · PRS · E; and (3) the effect of an environmental factor on an outcome varies depending on a binary covariate Xbinary as
In addition, we considered the condition where PRS has an effect on the assignment of an environmental factor, which would be often the case in observational studies as
where Xj is covariates with coefficients and CE is an intercept determined based on the prevalence of environmental factor, pE. While the simulation results under this condition were presented in the main text, those for scenarios where no association between PRS and an environmental factor were also shown in Supplementary Figs. 1 and 2. The expected treatment effect was calculated by dividing PRS values into small bins and taking the 1000 times average of the difference in the outcome trait value predicted from the model with an environmental factor of 1 and 0 in each PRS value.
We tested different values of each parameter as, n: {1000, 5000, 10,000, 50,000, 100,000}, pY: {0.1, 0.3}, pE: {0.1, 0.3}, aPRS: {1.25, 1.5, 2}, and aE or aE0: {1.25, 1.5, 2, 3}. The values for pY and pE represented low and high prevalence, as seen in diseases with lifestyle/behavioral risk factors and corresponding environmental factors. The values for aPRS were selected to represent higher and lower accuracy of PRS, with a value of 1.5 corresponding to typical accuracy, supported by mean values of 1.52 and 1.55 in the UKB and BBJ, respectively (Supplementary Table 1). The values for aE or aE0 were chosen to represent different magnitudes of environmental factor risks, compatible with those estimated in the real world, such as mean values of 2.89 for obesity and 1.35 for smoking in the UKB, and 1.26 for obesity and 1.99 for smoking in the BBJ (Supplementary Table 1). aE1 was set to 0.5, assuming situations where the effect of an environmental factor is halved depending on a binary covariate. bPRS was set to 1.2 considering a weaker effect of PRS on an environmental factor than on an outcome trait. The numbers of Xi and Xj were fixed at 10 and 5, respectively, with ai and bj set to 1.5.
Causal forest
We applied causal forest to build models that predict the ITE of environmental factors on the outcome trait. We included PRS and all covariates as inputs for the causal forest model. Then, heterogeneity in the predicted ITEs was evaluated across PRS values.
Linear regression analysis stratified by PRS values
We separated the entire data into ten groups according to PRS value. Then, we evaluated ATEs in each stratified group based on a linear regression model.
Linear regression analysis with an interaction term
We applied a linear regression model with an interaction term between PRS and environmental factor to the data, assuming that treatment effects can vary linearly according to PRS value.
Cohorts
UK Biobank
The UKB comprises health-related information ~500,000 individuals aged between 40 and 69 recruited from across the United Kingdom from 2006 to 201023. The process of patient registration, the GWAS data, and the quality control (QC) process are described elsewhere23. Briefly, we used the genomic data based on genotyping either by the Applied Biosystems UK BiLEVE Axiom Array or by the Applied Biosystems UK Biobank Axiom Array and imputation using a combination of the Haplotype Reference Consortium, UK10K and 1000 Genomes Phase 3 reference panels by IMPUTE4 software24. We included only individuals of British ancestry according to self-identification and criteria based on principal components (PCs)24. We excluded individuals of ambiguous sex (sex chromosome aneuploidy and inconsistency between self-reported and genetic sex), and outliers of heterozygosity or call rate of high-quality markers. We also excluded ≤2nd related samples (randomly selected samples to be remained) based on King’s kinship index > 0.088425.
The definition of cases and controls of T2D was based on ICD-10 codes and a previous study on diagnosis algorithms for diabetes in UKB26, defining individuals having a record of diagnosis of an ICD-10 code of E11 or probable T2D or possible T2D in the algorithms as cases and T2D unlikely in the algorithms as controls; we excluded individuals having a recode of diagnosis of ICD-10 codes of E10, E12, or E13 or probable type 1 diabetes, possible type 1 diabetes, or possible gestational diabetes in the algorithms. DL was defined as having a record with a primary or secondary diagnosis of ICD-10 codes of E78 or a medication history of cholesterol-lowering drugs. HTN was defined as having a record with a primary or secondary diagnosis of ICD-10 codes of I10–15, self-reported diagnosis of HTN, or a medication history of antihypertensive drugs. Incorporating medication histories into the definition of diseases can prevent missing hospital records and potential bias caused by the masking effect of drugs, although it involves a trade-off between this and the misclassification of non-diseased individuals as cases. CAD was defined based on a previous GWAS of CAD individuals with diabetes from UKB27, defining cases as individuals having a record of diagnosis of ICD-10 codes of I20–25, ICD-9 codes of 410–413, surgical intervention codes of K40–46, K49, K50, or K75, or self-reported diagnosis of angina pectoris or myocardial infarction. Obesity was defined as BMI > 30. Smoking and drinking histories were based on the UKB data-field codes of 20116 and 20117, respectively. For smoking history, current and previous smoking were combined into ever-smoker in the current study.
Although our main analysis targeted all cases that occurred before and during the observational period to maximize the sample sizes (i.e., case-control analysis as a part of cohort study), we additionally conducted a sensitivity analysis for individual risks evaluated in the UKB, exclusively including incidental cases. To achieve this, we defined cases diagnosed during and after the first round of assessment (i.e., 2006–2010) as baseline and incidental cases, respectively. Subsequently, baseline cases were excluded from the analysis. Incidental cases were determined solely based on the ICD diagnosis and a medication history to ensure a clear timeline. For the analysis using cardiovascular risk diseases as exposures, we excluded individuals who were recorded for these diseases (i.e., exposures) during the study period from unexposed groups to minimize the potential bias due to underdiagnosis at baseline. We randomly down-sampled the controls to have the same percentage of diseases as in the primary analysis.
BioBank Japan
The BBJ is a multi-institutional hospital-based registry that comprises DNA, serum, and clinical information of ~200,000 individuals of Japanese ancestry recorded from 2003 to 200728,29. The process of patient registration, the GWAS data, and the QC process are described in previous studies28,30,31. Briefly, the genomic data were based on genotyping with the Illumina HumanOmniExpressExome BeadChip or a combination of the Illumina HumanOmniExpress and HumanExome BeadChips and imputation with 1000 Genomes Project Phase 3 version 5 genotype and Japanese whole-genome sequencing data31,32. In the current study, individuals identified as non-Japanese either through self-reporting or as PC outliers from the East-Asian cluster were excluded33. We also excluded ≤2nd related samples (randomly selected samples to be remained) based on King’s kinship index > 0.088425. We used the cases of T2D, DL, HTN, and CAD, and smoking and drinking histories defined by the project29. The definition of ever- or never-smoker used in the current study was the same as that used in a previous GWAS on smoking status in BBJ34. In our main analyses, we used the same definition of obesity in the BBJ cohort as we did in the UKB cohort (i.e., BMI > 30). We also conducted the additional analyses using a lower cut-off point (i.e., BMI > 25) according to the definition of the Japan Society for the Study of Obesity, considering the specific distribution of BMI in the Japanese Asian population35. Individuals with any missing records were excluded. Given the high prevalence of diseases due to the unique recruitment approach in the BBJ (i.e., they enrolled participants with a diagnosis of at least 1 of 47 diseases)28,29, we randomly down-sampled the control to have the same percentage of diseases with the UKB so that the estimated exposure-related individual risk for each disease was comparable between the BBJ and UKB cohorts.
Calculation of PRSs
PRSs were calculated using a Bayesian PRS method, PRC-CS (auto mode)36, which has been shown to be superior to a conventional clumping and thresholding (C + T) method in robust benchmarking by aggregating the small risk effects from numerous variants with a continuous shrinkage36. A fully Bayesian approach of PRS-CS-auto does not require a validation dataset for tuning parameters; thus, we could use as much data as possible for the subsequent analysis. For calculation of PRSs, we only included SNPs with minor allele frequency > 0.01, average call rate > 0.98, Hardy–Weinberg equilibrium test P > 1.0 × 10−6, and INFO score > 0.8 in UKB and r2 imputation score > 0.5 in BBJ. We excluded palindromic and multi-allelic SNPs. We did not include sex chromosomes because of the controversy in using them for the calculation of PRSs22.
We preferentially used external ancestry-matched GWAS summary statistics that did not include each cohort. For phenotypes of which such external GWAS summary statistics were not publicly available, we constructed PRSs using a tenfold LOGO approach37. First, samples of each cohort were randomly split into ten groups. Then, we performed GWAS in each group by Plink38, meta-analyzed the GWAS results of nine groups in an inverse variance weighted method using Metal software39, and constructed PRSs for the remaining one using the result of the meta-analyzed GWAS. In a logistic model for GWAS in BBJ, we included age, sex, the top 10 PCs from the genotype data as covariates. The PRSs were normalized between the LOGO groups.
In UKB, we used external ancestry-matched GWAS summary statistics for T2D7 and CAD6, and used the LOGO approach for DL and HTN. In BBJ, external ancestry-matched GWAS summary statistics that do not include BBJ individuals are not publicly available for any diseases; we constructed PRSs using the LOGO approach.
Estimation of heterogeneity in the association between environmental risk factors and diseases in the biobank data
For each dataset from UKB and BBJ, we applied the causal forest model to build the models to predict the individual risk of diseases (CAD, T2D, DL, and HTN) associated with environmental risk factors (obesity and smoking)17. To note, we used the term exposure-related individual risk to differentiate it from ITE since the data available in the biobank were not interventional, but observational. In the causal forest approach, we included the following observable individual characteristics to the models: age, sex, PRS of the outcome disease, the top 10 PCs, and alcohol-drinking status. We also included the assessment center and genotyping array in UKB and the LOGO group in BBJ. In addition, we included obesity and ever-smoker as the characteristics in models in which exposure was not obesity and smoking, respectively. Because the causal model is designed to assess the heterogeneity in the association between a specific exposure and a specific outcome, we constructed individual models for each exposure of interest and outcome of interest. This approach means that the effects of multiple exposures were not evaluated simultaneously.
The same approach was applied to build the models to predict the individual risk of CAD associated with cardiovascular risk factors (T2D, DL, and HTN). Given the possible interaction between these risk factors for cardiovascular events40, we included all of them simultaneously in each model (i.e., model for T2D–CAD association, model for DL–CAD association, and model for HTN–CAD association). We also examined potential bias caused by not including them as covariates.
Ethics approval and consent to participate
We utilized only previously published publicly available biobank data; therefore, participant consent specific to this study was not required. This study was approved by the Ethical Committee of the Osaka University Graduate School of Medicine.
Statistics and reproducibility
Statistical analysis was conducted using R 3.6.1. A two-sided P value < 0.05 was considered statistically significant. The biobank GWAS genotype data were obtained as described in the “Data availability” section.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
Overview of this study
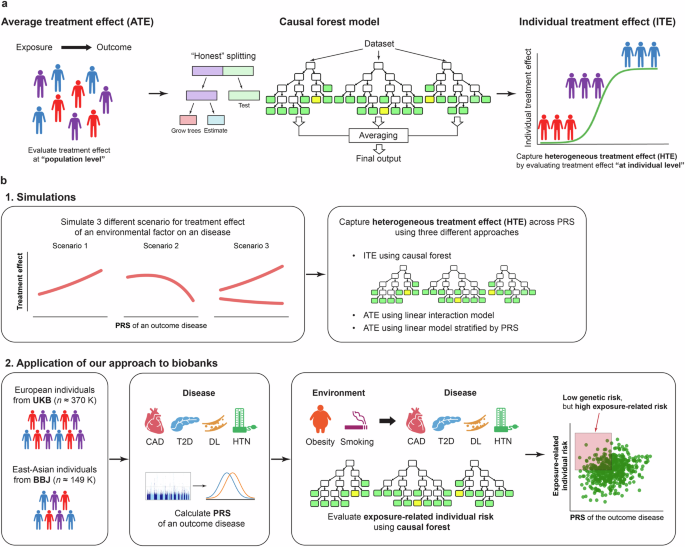
While outcome risk due to exposure is conventionally estimated for the entire study sample, recent machine-learning-based methods, such as causal forest, capture HTEs by predicting ITEs or individual disease risk related to exposure (Fig. 1a). First, we demonstrate this point with the illustration of the conceptual framework of estimating HTE across PRSs using simulated data representing PRS–ITE relationships under various conditions (Fig. 1b). We highlight potential limitations of conventional methods, which could be addressed by the machine-learning-based method. Second, we apply the causal forest method to biobanks to investigate the continuous relationship between PRSs and the estimated risk of cardiometabolic diseases associated with obesity and smoking (Fig. 1b).

a Conventionally, outcome risk due to an exposure is estimated for the entire study sample (i.e., at population level) although different individuals can have different treatment responses. Causal forest is a machine-learning method that enables us to capture heterogeneous treatment effects (HTEs) by predicting these individualized treatment effects (ITEs) or individual disease risk related to exposure, that can vary by individual characteristics. The estimated ITE represents the association between the environmental risk factors and the diseases in observational studies. Causal forest model uses random forests to partition the dataset into subpopulations with different magnitude of the risks according to observable characteristics. To minimize the bias, it also applies honest estimation in which the algorithm evaluates the results using out-of-sample. b. Our study consists of two main components: (1) simulations and (2) application of our approach to biobanks. In the simulations, we illustrated the conceptual framework of our study and demonstrated the methodological advancement of our approach using simulated data representing relationships between an exposure and an outcome disease under three different scenarios. In the application of our approach to biobanks, we separately analyzed European and East-Asian populations from UKB and BBJ, respectively. PRSs for individual diseases with lifestyle/behavioral risk factors (CAD, T2D, DL, and HTN) were calculated using publicly available GWAS summary statistics or a LOGO approach. Then, we estimated individual risk of diseases associated with environmental risk factors (obesity and smoking) using a causal forest model. We evaluated its heterogeneity across PRSs of the outcome diseases. To note, the red area in the scatter plot indicates individuals who are at low genetic risk, but at high disease risk associated with exposure (i.e., those who are suggested to be more likely to benefit from behavioral interventions to prevent the effect of the exposure). UKB UK Biobank, BBJ BioBank Japan, CAD coronary artery disease, T2D type 2 diabetes, DL dyslipidemia, HTN hypertension, PRS polygenic risk score.
Simulations for the conceptual clarification of evaluating heterogeneity via causal forest
We conducted Monte Carlo simulations for the varying relationships between PRSs and treatment effects of environmental factors on traits. We tested three different scenarios: (1) a simple model in which a binary outcome trait was determined by a logistic regression model of independent combinations of PRS values and an environmental factor, (2) a model complicated with an interactive effect between PRS and an environmental factor on the outcome trait, and (3) a binary outcome trait derived from either of two models with different effects of an environmental factor on the outcome depending on a binary covariate (e.g., sex). In simulations, we applied the causal forest approach to predict ITEs of an environmental factor, and then evaluated the correlation between PRS and ITEs. In addition, we investigated two conventional methods for evaluating average treatment effects (ATEs) that vary by a covariate (i.e., PRS in this case): (1) a linear regression model to estimate the risk difference in stratified groups according to PRS values and (2) a linear regression model with a linear interaction term between PRS and the target environmental factor. The simulations with a sample size of 100,000, disease prevalence of 0.1, environmental factor prevalence of 0.3, and specific values for other parameters are shown in the main text. While the results under the weak association between PRS and the environmental factor were presented here, those under no such association were shown in Supplementary Fig. 1.
In a simple model, the simulations revealed that there was a positive correlation between the expected treatment effect and PRS (Fig. 2a). However, the correlation was weak, which indicates that individuals with high PRS did not necessarily show a large magnitude of the treatment effect of environmental risk factors on diseases. Within a counterfactual framework, it is suggested that there would be a considerable number of individuals who are not at high genetic risk for the diseases, but more likely to benefit from interventions to prevent the environmental factor41.
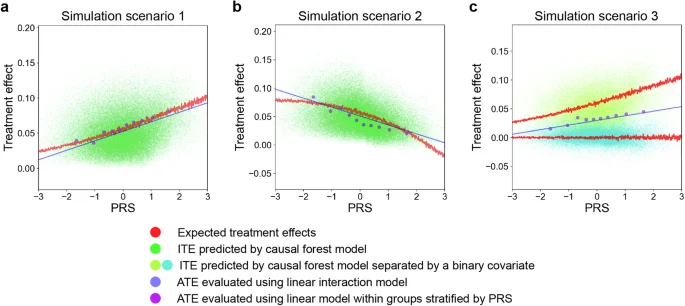
a–c Each panel represents a partial dependence plot of PRS on treatment effects of environmental risk factors on outcome traits under different simulation scenarios. The red lines represent the expected treatment effects from models used to generate the simulated data. The green (a, b) and yellow/cyan (c) dots represent the ITEs predicted by a causal forest model. In (c), the red lines are separated and dots are differently colored based on the binary covariate determining which models with different effects of the environmental factors generate them. The blue lines represent ATEs evaluated using a linear model with an interaction term between PRS values and environmental risk factors. The purple dots represent the ATEs evaluated with a linear model within groups stratified by PRS values. PRS polygenic risk score, ITE individualized treatment effect, ATE average treatment effect.
When there is an interaction effect between PRS and an environmental factor on the outcome trait, the PRS–ITE correlations varied and could even be negative depending on the interaction term values (Fig. 2b), which indicates the need to evaluate such relationship in each case. All methods successfully captured these relationships; however, ATEs evaluated using a linear interaction model deviated from the expected treatment effects.
When an outcome trait was derived from one of two different models depending on a covariate, the distributions of expected treatment effects across PRS varied by the covariate (Fig. 2c). This scenario is more likely in the real world, and the number of such covariates could be even more enormous. The causal forest successfully captured this distributional difference. On the other hand, stratified analysis by PRS failed to detect such differences. To note, stratification by the covariate could also capture the differences; however, it is unknown in advance which covariates should be used for stratification and the number of stratifications would be too enormous to be applied to real-world data. In addition, non-linear interactions in each stratified group would not be captured by linear models.
In the following, we used the best linear fit of the target estimand using the out-of-bag prediction to quantitatively evaluate the capability of causal forest to capture HTE complicated by different covariates as recommended in the general use of this algorithm (see “Methods” section)21. Its values significantly greater than 0 and close to 1 indicate that the model adequately captures heterogeneity (if any). We called it as the calibration coefficient here for simplicity. To gain insights for the practical applications of causal forest, we benchmarked its capability to capture HTE complicated by covariates with different values of each parameter (Supplementary Figs. 2 and 3). The causal forest performed better with larger sample sizes, suggesting that it would be more suitable for application to biobank-scale data. The model was not well calibrated when the number of cases and environmental factor assignments was insufficient, particularly requiring more than 50,000 samples with a prevalence of the outcome trait and environmental factor of 0.1. Additionally, it did not perform well when the treatment effect (i.e., degree of an environmental factor on the trait) itself was too low or high. To note, we need to carefully interpret model performance from these results because the degree of HTE itself also varies by different values of parameters.
An overview of the analysis for the biobank data
Here, we analyzed the relationships between environmental factors and outcome traits utilizing two nation-wide biobanks: UKB (n = 369,942) and BBJ (n = 149,421) (Fig. 1b). Since we used these real-world observational data within counterfactual framework, we herein applied the term of exposure-related individual risk instead of ITE to avoid strong causal statement for our estimand. For outcome diseases, we targeted CAD and its risk factor diseases, including T2D, DL, and HTN, because they are representative diseases with lifestyle/behavioral risk factors for which sufficient sample sizes were available in both biobanks. Regarding environmental factors, we targeted obesity (i.e., body max index (BMI) > 30 or not) and smoking status (i.e., ever-smoker or not) because they are two major factors for these diseases20. We did not target alcohol due to its highly variable and non-linear health burden depending on the amount of intake42, which complicates defining the exposure of interest and estimating its effect. The demographic characteristics of individuals are summarized in Supplementary Tables 2 and 3. PRSs of the diseases were calculated using publicly available GWAS summary statistics or a leave-one-group-out (LOGO) approach37. The PRSs demonstrated predictive ability in distinguishing cases from controls (Supplementary Fig. 4). The summary of the PRSs and LOGO GWAS are presented in Supplementary Tables 4 and 5, respectively. Then, we estimated individual risk of the diseases associated with the environmental risk factors using a causal forest model and evaluated its heterogeneity across the PRS values of the diseases and other characteristics.
The association between environmental risk factors and diseases varied by the PRSs
As shown in Fig. 3a, b, the individual risks of the diseases associated with obesity and smoking varied by the PRSs. The individual risk had positive correlation with PRSs, particularly for the relationship between obesity and T2D in the UKB (Spearman’s ρ = 0.61). This may suggest that individuals with high PRSs of T2D are more likely to benefit from behaviors that reduce the risk of obesity (e.g., physical activity and diet) than those with low PRSs in this cohort. In particular, individuals with PRSs > 90th percentile were estimated to have 2.3 times stronger risk of T2D associated with obesity than those with PRSs < 10th percentile (+17.7 percentage point [95% CI, 17.0–18.5] vs +7.60 percentage point [95% CI, 7.10–8.00]). However, such correlation was relatively weak in the BBJ cohort (Spearman’s ρ = 0.16); in contrast, the strongest positive correlation was observed at the relationship between smoking and CAD (Spearman’s ρ = 0.32). Thus, the correlation pattern between PRSs and disease risks associated with exposures varied across disease, environmental risk, and cohort. Notably, the correlations were weak or even negative for some environment–disease relationships (e.g., the relationships of smoking with DL and HTN and the relationship of obesity with HTN in the BBJ cohort). This indicates that individuals with high PRS did not necessarily show the large magnitude of the association between environmental risk factors and diseases; i.e., there would be a considerable number of individuals who are not at high genetic risk, but more likely to benefit from behavioral interventions41. For the BBJ cohort, we also reanalyzed the data using the population-specific definition of obesity (i.e., BMI > 25)35 to have adequate number of individuals in the exposed group so that we could capture the risk of obesity-related diseases in the Japanese population. Although the correlations between PRSs and individual risk of the diseases associated with the environmental risk factors became slightly stronger, they still remained weak, particularly for DL and HTN (Supplementary Fig. 5). In most of the environment–disease relationships, the models were well calibrated, with the calibration coefficients being nominally significant in 8 and 7 out of 8 relationships in the UKB and BBJ, respectively (P < 0.05). These significances were mostly preserved even after multiple test corrections, with 8 and 5 out of 8 relationships remaining significant in the UKB and BBJ, respectively (P < 0.05/8 = 0.00625, a Bonferroni-corrected P value threshold). However, the relationship between smoking and T2D in the BBJ cohort was poorly calibrated, probably due to the low association between smoking and T2D (P = 0.12; Fig. 3a, b).
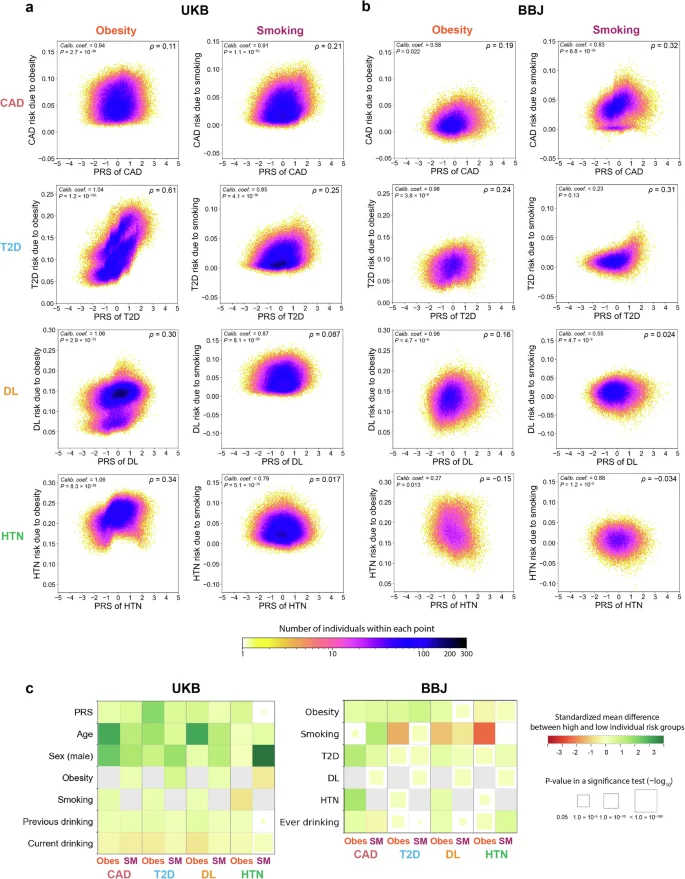
Each panel represents a partial dependence plot of PRS on the association between obesity or smoking and T2D, DL, HTN, or CAD in individuals from UKB (a) and BBJ (b). In each panel, the individual risks of disease associated with an environmental risk factor (the vertical axis) are shown along with the PRS values of a disease (the horizontal axis). The color of each dot represents the density of individuals within that dot according to the color bar at the bottom. We showed (i) calibration coefficients and their P values at the upper left and (ii) Spearman’s correlation coefficients between PRSs and disease risks associated with exposure at the upper right. c Comparison of characteristics (the vertical axis) between groups with high and low disease risks associated with each environmental risk factor (the horizontal axis) in the UKB and BBJ cohorts. In each panel, color of the squares represents the standardized mean differences in characteristics between the two groups. The size of squares corresponds to the P values of a two-tailed t-test for continuous characteristics and a Chi-square test for categorical characteristics between two groups. The size and color scales are provided on the right of the figure. The P values in all the panels were calculated from n = 369,942 samples in UKB. For BBJ, the P values were calculated from down-sampled data based on n = 149,421 independent samples to match the disease prevalence in the UKB. UKB UK Biobank, BBJ BioBank Japan, PRS polygenic risk score, CAD coronary artery disease, T2D type 2 diabetes, DL dyslipidemia, HTN hypertension, Obes obesity, SM smoking.
The correlation patterns between PRSs and exposure-related individual disease risks were complicated by individual characteristics such as age and sex
We further evaluated whether the observed pattern among the entire study sample could vary by individuals’ characteristics such as age and sex. When stratified by age, the individual risk of the diseases associated with environmental risk factors among older individuals was higher in the UKB cohort but lower in the BBJ cohort, particularly for the relationship between obesity and T2D (Supplementary Fig. 6). The correlation patterns between PRSs and disease risks associated with exposure were similar across ages in most cases (Supplementary Fig. 7). However, the correlation between the PRSs of HTN and the individual risks of HTN associated with obesity significantly varied by age in the UKB cohort (Spearman’s ρ = −0.30 and 0.47 among individuals aged ≥65 years and <65 years, respectively). There were also significant differences in the disease risks associated with exposures by sex (Supplementary Fig. 8); e.g., males were estimated to have 1.4 times stronger risk of T2D associated with obesity than females in the UKB cohort (+14.3 percentage point [95% CI, 13.9–14.6] vs +10.5 percentage point [95% CI, 10.3–10.7]). The correlations between PRSs and disease risks associated with obesity and smoking showed similar patterns regardless of sex (Supplementary Fig. 9). The sex differences in the correlations were less clear in the BBJ cohort (Supplementary Figs. 8 and 9).
Our ultimate goal is to identify groups for which behavioral changes to prevent environmental factors are more and less effective, which can be critical for in future personalized medicine. To this end, we compared the characteristics between individuals with high and low disease risks associated with environmental factors (>90th percentile and <10th percentile; Fig. 3c; Supplementary Tables 6 and 7). For example, in the UKB cohort, individuals with high disease risks associated with obesity and smoking were more likely to be older, male, and have higher PRS values than those with low disease risks associated with obesity and smoking. Although similar patterns were found except age in the BBJ cohort, individuals with high disease risks associated with obesity were more likely to be younger than those with low disease risks associated with obesity.
Sensitivity analysis for individual risks evaluated in the UKB
We additionally conducted a sensitivity analysis for individual risks evaluated in the UKB, exclusively including incidental cases. As shown in Supplementary Fig. 10, the calibration of causal forest was preserved (P < 0.05), despite a decrease in sample sizes. The overall correlation patterns between PRS and individual risks were consistent with those including all cases, as represented by the strong positive correlation in the relationship between obesity and T2D (Spearman’s ρ = 0.58).
Heterogeneous association of cardiovascular risk diseases with CAD across PRSs
T2D, DL, and HTN are major diseases that increase the risk of cardiovascular diseases; thus, we evaluated the individual risk of CAD associated with these cardiovascular risk diseases as exposures. While the highest positive correlation was found for the relationship between DL and CAD (Spearman’s ρ = 0.29 and 0.28 in the UKB and BBJ cohort, respectively), such correlation was not obvious for the relationship between T2D and CAD (|Spearman’s ρ| < 0.1 in both cohorts; Fig. 4a, b). In both cohorts, we found that individuals with high CAD risk associated with DL were more likely to be older, male, and smokers and had higher PRS values than those with low CAD risk associated with DL (Fig. 4c; Supplementary Tables 6 and 7).
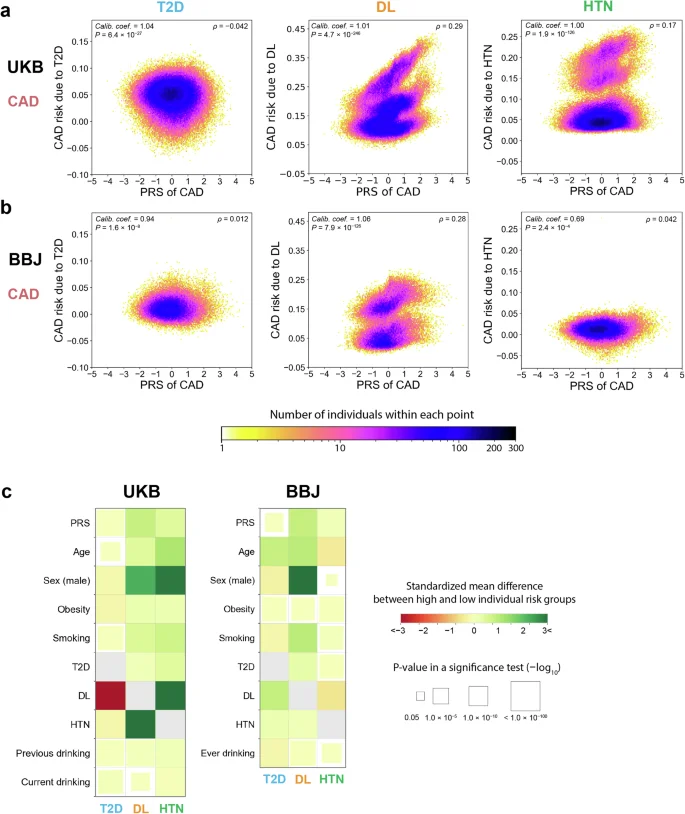
Each panel represents a partial dependence plot of PRS on the association between T2D, DL, or HTN and CAD in individuals from UKB (a) and BBJ (b). In each panel, the CAD risks associated with each cardiovascular risk factor (the vertical axis) are shown along with the PRS values for CAD (the horizontal axis). The color of each dot represents the density of individuals within that dot according to the color bar at the bottom. We showed (i) calibration coefficients and their P values at the upper left, and (ii) Spearman’s correlation coefficients between PRSs and disease risks associated with exposure and their P values at the upper right. c Comparison of characteristics (the vertical axis) between groups with high and low CAD risk associated with each cardiovascular risk factor (the horizontal axis) in the UKB and BBJ cohorts. In each panel, the color of squares represents the standardized mean differences in characteristics between the two groups. The size of squares corresponds to the P values of a two-tailed t-test for continuous characteristics and a Chi-square test for categorical characteristics between two groups. The size and color scales are provided on the right of the figure. The P values in all the panels were calculated from n = 369,942 samples in UKB. For BBJ, the P values were calculated from down-sampled data based on n = 149,421 independent samples to match the disease prevalence in the UKB. UKB UK Biobank, BBJ BioBank Japan, PRS polygenic risk score, CAD coronary artery disease, T2D type 2 diabetes, DL dyslipidemia, HTN hypertension.
In some cases, we found the difference in the distribution of one cardiovascular risk disease-related risk according to another, which would indicate the presence of interaction among these cardiovascular risk diseases40. For instance, when we stratified by DL status, individuals with DL were estimated to have 0.22 times lower risk of T2D-related CAD risk than those without DL in the UKB cohort (+1.38 percentage point [95% CI, 0.061–2.14] vs +6.25 percentage point [95% CI, 5.79–6.71]; P value for interaction between T2D and DL <0.001). In contrast, individuals with DL were estimated to have 3.3 times higher risk of HTN-related CAD risk than those without DL in the UKB cohort (+17.7 percentage point [95% CI, 17.0–18.4] vs +5.43 percentage point [95% CI, 5.26–5.61]; P value for interaction between HTN and DL <0.001). In addition, individuals with DL had a higher correlation between the CAD PRS and HTN-related CAD risk than those without DL (Spearman’s ρ = 0.39 vs 0.13). In the BBJ cohort, respective cardiovascular risk disease-related risks themselves were weaker and their interactive effects on the CAD risk and PRS were less obvious.
We performed the sensitivity analysis in the UKB for these relationships, exclusively including incidental cases. Specifically, the highest positive relationship between PRS and CAD risk due to DL was replicated (Spearman’s ρ = 0.20; Supplementary Fig. 11). The relationship for T2D could not be evaluated due to poor calibration (P > 0.05).
Lastly, while the results shown above are based on the models including risk diseases other than the target ones as covariates in the models, we performed an additional analysis not including them to demonstrate their potential impact on the results. The individual risks assessed in the models without these covariates exhibited higher magnitudes although the overall correlation patterns with PRS remained largely unchanged (Supplementary Fig. 12).
Discussion
Here, by utilizing the causal forest model and cross-population biobank resources, we demonstrated that the associations between environmental risk factors and diseases could vary by their PRSs, following the simulations showing the conceptual backgrounds of our study and the methodological advantages of the machine-learning-based approach. Particularly, we found that the higher PRSs of T2D were correlated with the stronger association between obesity and T2D in the UKB. However, such positive correlation was less clear for some of the other associations. These findings suggest that individuals with high PRSs will not necessarily benefit most from behavioral changes to prevent the effects of such environmental factors despite the usefulness of PRSs to predict individuals at high risk of diseases41. Given the heterogeneity in the disease risks associated with environmental factors across characteristics, identifying individuals with high benefit from behavioral changes as well as high genetic risk of diseases would help decision-makers to build the most efficient and effective precision preventive medicine approach to reduce global burden of diseases43,44. Because our study was based on the observational databases, the triangulation of evidence from other cohorts and several methodological approaches is required to establish the robustness of our findings and apply these concepts (i.e., targeting individuals with high benefit from behavioral changes) in clinical practice.
Although medicine often prioritizes individuals at high risk under an implicit assumption that such high-risk individuals receive high benefit from treatment, previous studies have suggested the risk of disease does not consistently correlate with the benefit of treatment44,45. Our study corroborates these findings, extending them to the genetic domain by demonstrating that individuals with elevated PRS may not derive the greatest benefit from environmental risk mitigation. Although the underlying mechanisms are not clear, the complex interplay of genetic factors, socioeconomic status, and disease history in this PRS-benefit discordance warrants deeper investigation, especially in the era of personalized medicine, where genetic insights guide treatment prioritization and resource allocation.
Previous studies well estimated the risk of environmental risk factors on CAD and cardiovascular risk factors in groups stratified by the values of PRSs10,11,12. For instance, a previous report showed that individuals with a higher genetic risk of CAD were more likely to benefit from lowering LDL cholesterol46, which is in line with our finding showing the positive correlation with PRS and CAD risk associated with DL. On the other hand, the current study provided several noteworthy progresses, facilitated by the methodological advantages of our approach. First, our approach could capture the heterogeneity in the association between environmental risk factors and diseases across continuous values of PRS without prior assumptions. By modeling together with other individual characteristics, we identified the heterogeneous patterns complicated by their multi-dimensional combinations. Of interest, the correlation between the HTN PRSs and obesity-related risks of HTN was reversed in older and younger individuals in the UKB cohort. Second, we obtained information that is practically useful when implementing targeted behavioral interventions by directly modeling exposure-related disease risk. As a result, some individuals even with low PRSs showed high disease risks associated with obesity and smoking, highlighting the importance of detecting such individuals to maximize the effectiveness of behavioral interventions. Considering that health burden of environmental risk factors could dynamically vary by other individual characteristics (e.g., age and sex), it is essential to identify high-treatment groups defined by such various characteristics. It is important to note that such implications should be carefully interpreted as they are based on the assumption that interventions work as expected for their target environmental factors in the population of interest.
Our finding of the positive correlation between PRSs and T2D risks associated with obesity in the UKB cohort is in line with a previous study showing the additive interaction of a healthy lifestyle (defined by diet, physical activity, smoking, alcohol intake, and BMI) and genetic risk score of T2D among European-ancestry adults12. A more recent study using the same cohort of US adults showed the consistent association between healthier diets and T2D regardless of genetic risk score, indicating that other lifestyle factors including physical activity and obesity might contribute to their original findings of interaction13. In addition, the risk and its correlation with the PRSs were small in the BBJ cohort, which might partially reflect the lower rate of obesity in East-Asian T2D patients47.
The correlation patterns for the relationship between other environmental risk factors and diseases were not consistent between the UKB and BBJ cohorts. First, we note that these differences might stem from the different schemes employed by the biobanks. Specifically, due to the hospital-based design of BBJ, wherein controls were selected from individuals with diseases other than the targets, the estimation of individual risks might be distorted compared to the general population-based design of UKB. Second, these results might represent the heterogeneity in the exposure–outcome associations depending on populations, which suggests the need for estimating the exposure-related individual risks according to target populations48. A possible explanation for the inconsistencies is that the proportion of genetic background associated with the effects of environmental risk factors within the overall PRS might vary across these populations. The PRS is an aggregate risk scale, and it is difficult to directly obtain biological insight. Therefore, the disentanglement of PRSs based on biological categories, such as pathways, may be an effective approach to infer the causes of differences and obtain biologically meaningful insight into the interaction between genetic and environmental risk factors. Nearly 90% of the disease-associated variants are within non-coding regions49, where in silico and in vitro functional annotations are massively in progress. Thus, such an annotation-based variant prioritization approach may be useful in the future.
In order to capture heterogeneity in exposure–outcome associations, it is fundamental to target cohorts with large sample sizes and various characteristics of individuals48,50. We addressed this point by utilizing the publicly available large-scale biobanks with individual genotype data. On the other hand, we could not rule out the possibility of reverse causation since clear time points of individual characteristics and medical conditions were often unavailable in the biobank data. To address this issue, we conducted additional analyses exclusively including incidental cases from the available UKB data, which yielded similar correlation patterns. Deciding whether to include all cases or focus solely on incidental cases involves balancing sample size sufficiency for calibration and evaluating heterogeneity against achieving stringent causation evaluation. Furthermore, in the current study, we focused on CAD, T2D, DL, and HTN, which had large sample sizes and known environmental risk factors. Although we attempted to evaluate the individual risks of other diseases—such as colorectal and lung cancer—associated with environmental factors, we could not well capture heterogeneous associations across PRSs due to their low heritability and relatively small sample sizes in the biobanks. We have several additional limitations and future advances to note. First, because causal forest is not a statistical tool to address the systematic biases occurring in observational studies, our results might have suffered from bias due to unmeasured confounding. As we examined the potential effects of including confounding covariates or not into the models, this could affect the magnitude or relationships to PRS of exposure-related risks. Second, our findings of the heterogeneity by age should be carefully interpreted because age was measured at the study enrollment and might not be related to exposure and outcome status. Third, exposures were self-reported, and thus these variables might have been misclassified51. Fourth, we treated the exposures as binary; however, their internal distributions (e.g., severity of obesity, pack-years of smoking, etc.) may vary according to different characteristics. On the other hand, we note that interpreting and generalizing the effects of changes in environmental factors by treating them as continuous variables could be intrinsically challenging, as these effects may vary depending on their baseline values (e.g., 5 kg/m2 increase in BMI would have different impacts on health for people with BMI of 15 kg/m2 compared to those with BMI of 30 kg/m2). Fifth, the LOGO approach could cause potential biases, such as overfitting to the target cohorts, compared to employing GWAS sum stats from external cohorts and heterogeneity in PRS compositions across different folds. Sixth, given the hospital-based design of BBJ, evaluating general population-based biobanks for the Japanese population is necessary to robustly obtain shared and distinct insights into exposure-related risks across PRSs between populations. Lastly, given the different patterns of the heterogeneous association between environmental factors and diseases across the cohorts in our study, our results may not be generalizable to other populations, emphasizing the need to individually evaluate them depending on specific purposes.
Conclusion
To the best of our knowledge, this study first introduced the concepts of predicting exposure-related risks at the individual level and evaluating the potential heterogeneity in the individual risks across PRSs using the machine-learning-based approach. While PRSs can be useful in identifying high-risk groups for diseases, they may not necessarily provide direct information in selecting individuals who are more effective for preventive behavioral interventions. Predicting not only high-risk groups but also those who are more likely to benefit from treatment may maximize the performance of precision medicine using genetic data that should be the subject of future research.
Data availability
GWAS data of the BBJ are available at the NBDC Human Database (Research ID: hum0014). Access to the UK Biobank data can be obtained by applying through the UK Biobank Access Management System, as detailed at https://www.ukbiobank.ac.uk/. We obtained the UKB GWAS data via application number 47821. The source data for Figs. 3c and 4c are available in Supplementary Tables 6 and 7.
Code availability
R scripts to perform the HTE analysis used in this study are shared in a GitHub repository (https://github.com/tatsuhikonaito/PRS_HTE)52.
References
-
Vos, T. et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 396, 1204–1222 (2020).
Google Scholar
-
World Health Organization. Cardiovascular diseases (CVDs) https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (2021).
-
Bennett, J. E. et al. NCD Countdown 2030: worldwide trends in non-communicable disease mortality and progress towards Sustainable Development Goal target 3.4. Lancet 392, 1072–1088 (2018).
Google Scholar
-
Ramaswami, R., Bayer, R. & Galea, S. Precision medicine from a public health perspective. Annu. Rev. Public Health 39, 153–168 (2018).
Google Scholar
-
Wongvibulsin, S., Martin, S. S., Saria, S., Zeger, S. L. & Murphy, S. A. An individualized, data-driven digital approach for precision behavior change. Am. J. Lifestyle Med. 14, 289–293 (2020).
Google Scholar
-
Nikpay, M. et al. A comprehensive 1000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat. Genet. 47, 1121–1130 (2015).
Google Scholar
-
Scott, R. A. et al. An expanded genome-wide association study of type 2 diabetes in Europeans. Diabetes 66, 2888–2902 (2017).
Google Scholar
-
Adeyemo, A. et al. Responsible use of polygenic risk scores in the clinic: potential benefits, risks and gaps. Nat. Med. 27, 1876–1884 (2021).
Google Scholar
-
Lewis, C. M. & Vassos, E. Polygenic risk scores: from research tools to clinical instruments. Genome Med. 12, 44 (2020).
Google Scholar
-
Khera, A. V. et al. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N. Engl. J. Med. 375, 2349–2358 (2016).
Google Scholar
-
Abdullah Said, M., Verweij, N. & Van Der Harst, P. Associations of combined genetic and lifestyle risks with incident cardiovascular disease and diabetes in the UK biobank study. JAMA Cardiol. 3, 693–702 (2018).
Google Scholar
-
Ding, M. et al. Additive and multiplicative interactions between genetic risk score and family history and lifestyle in relation to risk of type 2 diabetes. Am. J. Epidemiol. 189, 445–460 (2020).
Google Scholar
-
Merino, J. et al. Polygenic scores, diet quality, and type 2 diabetes risk: an observational study among 35,759 adults from 3 US cohorts. PLoS Med. 19, e1003972 (2022).
Google Scholar
-
Dahabreh, I. J. & Kazi, D. S. Toward personalizing care. JAMA 329, 1063 (2023).
Google Scholar
-
Powers, S. et al. Some methods for heterogeneous treatment effect estimation in high dimensions. Stat. Med. 37, 1767–1787 (2018).
Google Scholar
-
Athey, S. & Imbens, G. Recursive partitioning for heterogeneous causal effects. Proc. Natl. Acad. Sci. USA 113, 7353–7360 (2016).
Google Scholar
-
Wager, S. & Athey, S. Estimation and inference of heterogeneous treatment effects using random forests. J. Am. Stat. Assoc. 113, 1228–1242 (2018).
Google Scholar
-
Athey, S. & Wager, S. Estimating treatment effects with causal forests: an application. Obs. Stud. 5, 37–51 (2019).
Google Scholar
-
Inoue, K., Seeman, T. E., Horwich, T., Budoff, M. J. & Watson, K. E. Heterogeneity in the association between the presence of coronary artery calcium and cardiovascular events: a machine-learning approach in the MESA study. Circulation 147, 132–141 (2023).
Google Scholar
-
Nyberg, S. T. et al. Association of healthy lifestyle with years lived without major chronic diseases. JAMA Intern. Med. 180, 760 (2020).
Google Scholar
-
Chernozhukov, V., Demirer, M., Duflo, E. & Fernández-Val, I. Generic Machine Learning Inference on Heterogeneous Treatment Effects in Randomized Experiments, with an Application to Immunization in India https://www.nber.org/papers/w24678, https://doi.org/10.3386/w24678 (NBER Working Paper, 2018).
-
Choi, S. W., Mak, T. S. H. & O’Reilly, P. F. Tutorial: a guide to performing polygenic risk score analyses. Nat. Protoc. 15, 2759–2772 (2020).
Google Scholar
-
Sudlow, C. et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
Google Scholar
-
Bycroft, C. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209 (2018).
Google Scholar
-
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010).
Google Scholar
-
Eastwood, S. V. et al. Algorithms for the capture and adjudication of prevalent and incident diabetes in UK Biobank. PLoS ONE 11, e0162388 (2016).
Google Scholar
-
Fall, T., Gustafsson, S., Orho-Melander, M. & Ingelsson, E. Genome-wide association study of coronary artery disease among individuals with diabetes: the UK Biobank. Diabetologia 61, 2174–2179 (2018).
Google Scholar
-
Nagai, A. et al. Overview of the BioBank Japan Project: study design and profile. J. Epidemiol. 27, S2–S8 (2017).
Google Scholar
-
Hirata, M. et al. Cross-sectional analysis of BioBank Japan clinical data: a large cohort of 200,000 patients with 47 common diseases. J. Epidemiol. 27, S9–S21 (2017).
Google Scholar
-
Kanai, M. et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat. Genet. 50, 390–400 (2018).
Google Scholar
-
Akiyama, M. et al. Characterizing rare and low-frequency height-associated variants in the Japanese population. Nat. Commun. 10, 4393 (2019).
Google Scholar
-
Okada, Y. et al. Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat. Commun. 9, 1–10 (2018).
Google Scholar
-
Akiyama, M. et al. Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat. Genet. 49, 1458–1467 (2017).
Google Scholar
-
Matoba, N. et al. GWAS of smoking behaviour in 165,436 Japanese people reveals seven new loci and shared genetic architecture. Nat. Hum. Behav. 3, 471–477 (2019).
Google Scholar
-
The Examination Committee of Criteria for ‘Obesity Disease’ in Japan. New criteria for ‘obesity disease’ in Japan. Circ. J. 66, 987–992 (2002).
Google Scholar
-
Ge, T., Chen, C. Y., Ni, Y., Feng, Y. C. A. & Smoller, J. W. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 10, 1–10 (2019).
Google Scholar
-
Sakaue, S. et al. Trans-biobank analysis with 676,000 individuals elucidates the association of polygenic risk scores of complex traits with human lifespan. Nat. Med. 26, 542–548 (2020).
Google Scholar
-
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Google Scholar
-
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010).
Google Scholar
-
Hu, G., Jousilahti, P. & Tuomilehto, J. Joint effects of history of hypertension at baseline and type 2 diabetes at baseline and during follow-up on the risk of coronary heart disease. Eur. Heart J. 28, 3059–3066 (2007).
Google Scholar
-
Rothman, K. J. & Greenland, S. Causation and causal inference in epidemiology. Am. J. Public Health 95, S144–S150 (2005).
Google Scholar
-
Klatsky, A. L. Alcohol and cardiovascular diseases: where do we stand today? J. Intern. Med. 278, 238–250 (2015).
Google Scholar
-
Li, Z., Chen, J., Laber, E., Liu, F. & Baumgartner, R. Optimal treatment regimes: a review and empirical comparison. Int. Stat. Rev. https://doi.org/10.1111/insr.12536 (2023).
-
Inoue, K., Athey, S. & Tsugawa, Y. Machine-learning-based high-benefit approach versus conventional high-risk approach in blood pressure management. Int. J. Epidemiol. 52, 1243–1256 (2023).
Google Scholar
-
Cheung, L. C., Berg, C. D., Castle, P. E., Katki, H. A. & Chaturvedi, A. K. Life-gained–based versus risk-based selection of smokers for lung cancer screening. Ann. Intern. Med. 171, 623 (2019).
Google Scholar
-
Natarajan, P. et al. Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation 135, 2091–2101 (2017).
Google Scholar
-
Ma, R. C. W. & Chan, J. C. N. Type 2 diabetes in East Asians: similarities and differences with populations in Europe and the United States. Ann. N. Y. Acad. Sci. 1281, 64–91 (2013).
Google Scholar
-
Bryan, C. J., Tipton, E. & Yeager, D. S. Behavioural science is unlikely to change the world without a heterogeneity revolution. Nat. Hum. Behav. 5, 980–989 (2021).
Google Scholar
-
Edwards, S. L., Beesley, J., French, J. D. & Dunning, A. M. Beyond GWASs: illuminating the dark road from association to function. Am. J. Hum. Genet. 93, 779–797 (2013).
Google Scholar
-
Kent, D. M., Steyerberg, E. & van Klaveren, D. Personalized evidence based medicine: predictive approaches to heterogeneous treatment effects. BMJ 363, k4245 (2018).
Google Scholar
-
Xue, A. et al. Genome-wide analyses of behavioural traits are subject to bias by misreports and longitudinal changes. Nat. Commun. 12, 20211 (2021).
Google Scholar
-
Naito, T. Machine learning reveals heterogeneous associations between environmental factors and cardiometabolic diseases across polygenic risk scores. https://github.com/tatsuhikonaito/PRS_HTE, https://doi.org/10.5281/zenodo.11266192 (2024).
Acknowledgements
We would like to thank all the participants involved in this study. K.I. was supported by JSPS KAKENHI (22K17392), the Japan Agency for Medical Research and Development (AMED; JP22rea522107), and the Program for the Development of Next-generation Leading Scientists with Global Insight (L-INSIGHT), sponsored by the Ministry of Education, Culture, Sports, Science and Technology (MEXT). Y.O. was supported by JSPS KAKENHI (22H00476), and AMED (JP22ek0410075, JP23km0405211, JP23km0405217, JP23ek0109594, JP23ek0410113, JP223fa627002, JP223fa627010, JP233fa627011, JP23zf0127008), JST Moonshot R&D (JPMJMS2021, JPMJMS2024), Takeda Science Foundation, Bioinformatics Initiative of Osaka University Graduate School of Medicine, Institute for Open and Transdisciplinary Research Initiatives, Center for Infectious Disease Education and Research (CiDER), and Center for Advanced Modality and DDS (CAMaD), Osaka University. S.N. was supported by AMED (JP24tm0424228), Takeda Science Foundation, and Japan Foundation for Applied Enzymology.
Author information
Authors and Affiliations
Consortia
Contributions
T.N., K.I., and Y.O. designed the study. T.N. and K.I. conducted the data analysis and wrote the manuscript. S.N., K.S., N.K., T.T., T.Y., and T.K. managed the project and data collection. BBJ and K.M. managed the samples and provided the data. Y.O. supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Medicine thanks Masao Iwagami, Qingpeng Zhang and the other, anonymous, reviewer for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplemental Information
Reporting Summary
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
Reprints and permissions
About this article
Cite this article
Naito, T., Inoue, K., Namba, S. et al. Machine learning reveals heterogeneous associations between environmental factors and cardiometabolic diseases across polygenic risk scores.
Commun Med 4, 181 (2024). https://doi.org/10.1038/s43856-024-00596-7
-
Received: 12 October 2023
-
Accepted: 22 August 2024
-
Published: 20 September 2024
-
DOI: https://doi.org/10.1038/s43856-024-00596-7