Abstract
Chemical space modelling has great importance in unveiling and visualising latent information, which is critical in predictive toxicology related to drug discovery process. While the use of traditional molecular descriptors and fingerprints may suffer from the so-called curse of dimensionality, complex networks are devoid of the typical drawbacks of coordinate-based representations. Herein, we use chemical space networks (CSNs) to analyse the case of the developmental toxicity (Dev Tox), which remains a challenging endpoint for the difficulty of gathering enough reliable data despite very important for the protection of the maternal and child health. Our study proved that the Dev Tox CSN has a complex non-random organisation and can thus provide a wealth of meaningful information also for predictive purposes. At a phase transition, chemical similarities highlight well-established toxicophores, such as aryl derivatives, mostly neurotoxic hydantoins, barbiturates and amino alcohols, steroids, and volatile organic compounds ether-like chemicals, which are strongly suspected of the Dev Tox onset and can thus be employed as effective alerts for prioritising chemicals before testing.
Introduction
The canonical representation of chemical spaces based on a coordinate system with multiple dimensions suffers from several issues. It is not invariant to the chosen representation: changing the adopted features can dramatically affect the boundaries of the chemical space and its properties. It cannot deal naturally with features that are both discrete and continuous1,2. In this regard, metric spaces can make things even harder while complex networks, which are intrinsically non-metric, can promptly offer a solution.
In recent years, the opportunities given by the adoption of complex networks to model the chemical spaces, the so-called chemical space networks (CSNs), have been widely investigated. Several fields have been studied, such as medicinal chemistry, physicochemical properties, and de novo drug design, just to mention a few3,4,5,6,7,8. An additional advantage provided by CSNs is the smart mathematical framework behind them that is the graph theory. Centrality metrics such as degree, betweenness and eigenvector centrality can suitably characterise the behaviour of the chemicals within a network, while their distribution can deepen our understanding of the network organisation and, therefore, of the resulting chemical space9. Topological properties allow for the characterization of a network organisation, for example the presence of hubs or communities. Features, such as scale-freeness or small-worldness, can signal the presence of patterns and dynamics within a network as extensively reported elsewhere10,11; by contrast, random graph models, such as the Erdos–Renyi (ER) model, can be used for benchmarking or to assess the meaningfulness of specific structures and architectures12.
Previous studies have investigated CSNs as threshold networks, i.e., networks whose structures depend and vary according to specific cut-off values set on the network connections. The constituent elements of these networks, usually called nodes, are chemicals while connections are pairwise molecular similarities: these studies were aimed at comparing different datasets13,14,15. This approach has shown how different similarity metrics generate different CSNs, how different choices of the similarity cut-off affect nodal properties like degree or assortativity, among the others, and the presence of molecular communities16,17,18. Defining an optimal cut-off is far from being a simple fine-tuning matter and varying the similarity threshold adopted to construct a network deeply shapes the network topology and its meaningfulness14,19,20. Moreover, the task is complicated by the huge heterogeneity of the chemical space. To mitigate this issue, this work will be focused on a reduced yet extremely interesting class of chemicals, consisting of small molecules experimentally labelled as toxic with respect to developmental toxicity (Dev Tox).
Herein, we investigate the possibility of choosing an optimal threshold based on statistical mechanics properties. We identify a first-order phase transition, a signal of emergent behaviours within a complex system, as a flag that an optimal cut-off has been reached. Although this perspective has been thoroughly reported in several case studies and has demonstrated its effectiveness by providing fundamental advances in our understanding of collective phenomena21,22,23,24,25,26, an application to the CSNs is still missing. More broadly, our goal is to provide interpretable insights on CSNs.
The data investigated deal with Dev Tox. This concerns offspring abnormal development due to the exposure to harmful agents or to hazard conditions27. It is a complex human health endpoint, of utmost importance especially for the care of the maternal and child health. Predicting the Dev Tox onset remains extremely challenging and far from reaching satisfactory levels of accuracy28,29. In the last decade, several predictive approaches, especially based on machine learning, have been proposed30,31,32,33.
The knowledge gap in understanding Dev Tox depends on two intimately related aspects: on one side the limited amount of Dev Tox measured data as well as their uncertainty and on the other the structural complexity of the chemicals reflecting the space heterogeneity. Hence, we propose to employ the CSN perspective to model such heterogeneity and gain fundamental insights about which toxic chemicals share common molecular patterns and, eventually, deepen our rational understanding of the latent toxicological mechanisms behind Dev Tox. In this respect, our study also falls in the broad cutting-edge domain of eXplainable Artificial Intelligence (XAI)34,35,36,37.
Results
Criticality signals optimal thresholding
To study the CSN of Dev Tox, a database of small molecules, whose toxic effects are well established, was collected. To the best of our knowledge, the data used here represent the largest publicly available base of knowledge for Dev Tox. Two main sources were taken into account for modelling Dev Tox. The CAESAR30 and the Procter & Gamble (P&G) datasets38, including 201 and 621 experimentally toxic chemicals, respectively. The Food and Drug Administration (FDA) classifies as toxic the chemicals belonging to one of the three following categories39,40: class C that reports chemicals tested positive for Dev Tox in animal studies; class D that reports chemicals tested positive for Dev Tox only in human studies; class X that reports chemical tested positive in both animal and human studies and/or had evidence of foetal risk based on human experience.
Preliminary examinations were carried out to select the most reliable chemicals for Dev Tox modelling and, after removing duplicates, a number N of 684 toxic chemicals were included in this study. List of all chemicals with their structures and Dev Tox annotations is available in the Table S1 of the Supporting Information. The Table S1 also includes a list of 135 non-toxic chemicals available from the previously mentioned data repository which were used for classification purposes.
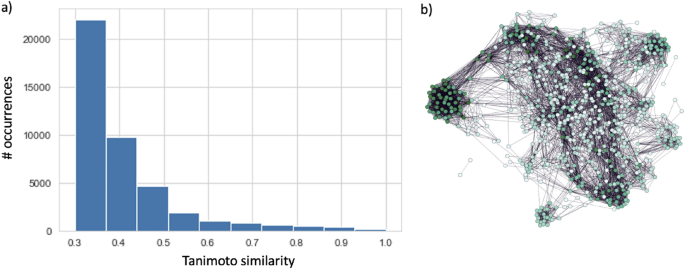
N(N-1)/2 pairwise Small Molecule Subgraph Detector (SMSD)41 Tanimoto42 similarity measures were calculated and connections between nodes with similarity greater than the cut-off value, set to 0.3, were established based on the assumption that a very low similarity does not reflect any meaningful information. Such a choice allowed a remarkable computational burden reduction. The resulting similarity distribution along with the CSN is presented in Fig. 1.

Similarity distribution of Tanimoto values (a). Chemical space network (b).
This study aims at analysing the CSN topology, thus network weights are neglected as topological features are generally weight-independent43,44; nevertheless, weights become crucial when considering different cut-off values and this can dramatically affect topology. Hence, by thresholding Tanimoto similarities at different cut-off values, we investigated the variations occurring within the CSN in terms of three main centrality metrics: degree (d), betweenness (b) and eigenvector centrality (e). Moreover, a paired analysis on an ensemble of ER graphs comparable with the CSN was carried out.
For each threshold value, the number of CSN edges (E) was computed with the maximum possible value being:
with N, the number of chemicals, being the CSN order.
Thus, the connection probability
was calculated and an ER model (Gleft(N,pright)) was simulated (for statistical robustness, 20 different simulations were performed). The results are shown in Fig. 2.

Centrality metrics as a function of the Tanimoto similarity for both (a) CSN and (b) ER networks. Betweenness is represented in black diamonds, degree with blue crosses and eigenvector centrality with red triangles. Centrality measures are scaled in the [0,1] interval for ease of comparison.
The number of CSN connections occurring in the model ranges from 0 (when the Tanimoto threshold is 1) to 41,807 (when the Tanimoto threshold is 0.3) while the resulting connection probability p ranges approximately within (0div {10}^{-1}) which includes the ER critical probability, which is ({p}_{crit} sim 1/N={10}^{-3}). As expected, centrality metrics intrinsically related to edge counting, such as degree and eigenvector centrality, showed monotonically decreasing trends. This can easily be explained as a direct consequence of the edge removal. Apart from statistical fluctuations, the degree is equal for both models. In fact, by construction, the ER model is simulated with the connection probability retrieved by the CSN. Eigenvector centrality has higher values in the ER ensemble, while for higher probability values the two trends are substantially similar. An analogous consideration holds for eigenvector centrality and betweenness (see Fig. S1 of the Supporting Information for a detailed comparison). For both CSN and ER models, betweenness increases with connection probability until a maximum is reached, then betweenness abruptly decreases. Interestingly, while this phase transition occurs as expected with (p sim {p}_{crit}) in the ER model, for the CSN model this phase transition occurs at a slightly higher value ({p}_{crit}^{CSN} {sim 5cdot 10}^{-3}) which corresponds to a Tanimoto similarity of ~ 0.7. Overall, these results highlight the presence of an optimal cut-off signalled by the behaviour of betweenness and a problematic similarity between the constructed CSN and a random graph.
The CSN is not random
The CSN being a random graph would be not only far from intuition, as chemicals showing similar behaviours should be close in the chemical space, but it would also pose fundamental issues as a random network by definition does not include meaningful structures.
It can be easily shown that the phase transition signalled by the peak in betweenness corresponds to the first-order phase-transition of the giant component in a random graph, although it does not occur at (p 1/N), see Fig. 3. The fraction of nodes within the giant component becomes non-null at the same critical probability ({p}_{crit}) at which betweenness abruptly increases (as previously shown in Fig. 2). However, the studied CSN cannot be a random graph and the definitive proof is provided by assortativity. In fact, Fig. 3 shows that assortativity increases with the connection probability until it reaches a maximum at ({p}_{crit}).

Giant component phase transition. The percentage of nodes within the giant component (black crosses) and the normalised assortativity (blue points) are shown as functions of connection probability.
This behaviour is a consequence of the transitivity of Tanimoto similarity. Low probabilities correspond to high similarity; therefore, if two chemicals are connected to a third one then they will probably be connected. This is a typical assortative behaviour, and it is not consistent with a random graph whose assortativity should be close to zero. In fact, the CSN assortativity dramatically drops, when more and more edges are added, tending to the behaviour of a random graph. Thus, despite thresholding the CSN at criticality yielded the emergence of a giant component, the meaningfulness of CSN inner structures, based on molecular similarity, is preserved.
The Dev Tox archetypes
At criticality, the CSN experiences the giant component formation while satellite groups of few chemicals are also present. A community detection analysis was performed along with a modularity analysis to highlight the inner CSN organisation; moreover, the community cardinality was examined, see Fig. 4.

CSN at criticality: the largest 8 communities are outlined with different colours. The network nodes at criticality are basically scattered among several communities, the first 30 communities are shown here. The panel confirms that at criticality the fraction of isolated nodes is reasonable while the partition quality, in terms of modularity, reaches more than satisfactory levels.
At criticality, almost 25% of network nodes are grouped within the top three populated communities, consisting of 46, 45 and 42 elements. The overall modularity and the percentage of connected nodes exceeded 80%. These findings suggest the existence of specific communities, based on molecular patterns, that we will call Dev Tox “archetypes”. It should be noted that these archetypes should not be strictly intended as toxicological classes; in fact, by construction, they are based only on structural similarities of toxicophores evaluated by means of the Tanimoto metric. To gain further insights into the chemical meaning of these communities, we computed hundreds of molecular descriptors (from physicochemical to auto-correlation properties) for each toxicophore and investigated their statistical association within the archetypes. After Bonferroni correction, we found 145 descriptors whose distributions can be significantly distinguished, at 1% significance, within the three communities.
This analysis highlighted the presence of descriptors capturing basic and easy-to-interpret features such as molecular weight, number of valence electrons and molecular refractivity. Interestingly, such descriptors are of particular interest when evaluating properties of fundamental importance such as drug-likeness45,46. Other descriptors significantly related to communities were well-known topochemical indices such as BCUT descriptors, BertzCT and molecular connectivity chi indexes47. Finally, an extremely relevant role was played by Moreau-Broto autocorrelation descriptors48. Topological autocorrelation is frequently used in Quantitative Structure–Activity Relationship (QSAR) models49 to assess how specific physicochemical properties are spatially distributed along molecules. Here, almost 50% of significant descriptors consisted of autocorrelation patterns, specifically involved with (i) atomic properties (number of valence or sigma electrons), mass, atomic numbers; (ii) electronegativity (Sanderson, Pauling and Allred-Rochow)50.
Besides statistical significance, the median value for each descriptor and its interquartile range were evaluated to highlight the different behaviour within each community along with its variability. For example, the molecular refractivity is shown in Fig. 5.

The boxplot shows the variability range for the molecular refractivity within the top three populated communities.
A comprehensive list of significant descriptors and their characteristic ranges for Dev Tox is provided in the Table S2 of the Supporting Information. These results confirm that the communities reflect structural similarities and therefore they include chemicals with different properties.
In particular, the first community includes heterogeneous toxicophores in terms of structural moieties, being this probably due to its large size. In general, it includes aryl derivatives mainly comprising barbiturates, hydantoins and amino alcohols, commonly used as anticonvulsant drugs, GABA modulators, excitatory amino acid antagonists, hypnotic and sedative drugs, see Fig. 6a. The second community cover toxicophores with well-known cyclopentanoperhydrophenanthrene cores typical of steroids, responsible of essential biological functions such as fluidity and permeability regulation also known for fertility impairment, see Fig. 6b. The third community is made by small Volatile Organic Compounds (VOCs) ether-like chemicals, which act as pollutants and food toxins, see Fig. 6c.

The most representative chemical structures of the three top communities: (a) aryl derivatives mainly comprising barbiturates and amino alcohols; (b) cyclopentanoperhydrophenanthrene cores typical of the steroid lipid family; (c) small Volatile Organic Compounds (VOCs) ether-like chemicals.
Finally, we explored whether the toxicophores within a community shared specific drug targets generally consisting of receptor and enzyme proteins. To this aim, we used the PLATO platform51, which is specifically aimed at pairing biological targets to small molecules on the basis of the calculated similarity with respect to known ligands annotated with experimental biological measures retrieved from the CHEMBL database. Based on highly occurring targets, we found that the first community engage mostly targets relevant for the central nervous system, normally engaged by hypnotic, sedative and anticonvulsant drugs. While the community of steroids mainly interplayed with the hormonal system including mineralocorticoid and glucocorticoid receptors, progesterone receptors and androgen-binding protein receptors. The third community, mostly composed of VOCs ethers-like chemicals, did not pair with any verified target and this could be due to their low similarity to known drugs.
The CSN predictive power
To evaluate the potential of CSN as a support for predictive investigations, a further analysis was carried out. We included in the CSN the nodes representing the non-toxic chemicals listed in Table S1 and designed a simple classification framework to assess to what extent the CSN is able to distinguish toxic from non-toxic chemicals. For each node/chemical to be classified, the connected nodes/chemicals were inspected along with their pairwise Tanimoto similarities; then, the classification score was computed with a weighted average. The adopted weights were the computed similarities so that the most similar chemicals were the most influential in classification. Classification results in terms of accuracy, sensitivity, specificity and f1 metrics are presented in Fig. 7.

Classification metrics for CSN predictive power as a function of the Tanimoto similarity. At the preferred Tanimoto similarity threshold of ~ 0.7, the model achieves a good overall accuracy (~ 80%) with great sensitivity (> 90%) but poor specificity (~ 25%).
For the sake of completeness, the classification results explored the whole range of possible Tanimoto similarity thresholds. At the critical threshold (~ 0.7), the model was able to achieve a good overall accuracy (~ 80%) and f1 (~ 90%) an extremely high sensitivity (> 90%) while specificity was significantly lower (~ 25%). Performance uncertainties were obtained by means of an 80% hold-out cross-validation, iterated 100 times.
Discussion
The study of the chemical space is fundamentally based on one basic assumption: the structure of chemicals yields information about its physicochemical and biological properties, including the pharmacological or toxicological behaviour52,53,54,55,56. Although this paradigm is generally accepted and it has been verified for several applications, some important issues still remain not completely solved56,57,58,59,60,61. A fundamental limitation to this perspective concerns the extreme variability of physicochemical properties even when few or just one atomic variation occurs within a molecular structure. Hence, a potentially disrupting consideration arises: if even a subtle molecular change can turn a toxic chemical into a non-toxic chemical, then any conclusion drawn from molecular similarity is uninformative, the chemical space network and its inner communities are meaningless. A corollary question, which would prevent any possible further consideration, concerns the possibility of uniquely defining a scale at which to investigate the chemical space, i.e., to identify a suitable threshold for molecular similarity measurements.
Here, we provide an answer to both questions and demonstrate how they are indissolubly related. In fact, our findings showed an outstanding overlap between the Dev Tox CSN and an ER model; this was observed at all the threshold values, thus dangerously suggesting the possibility that the CSN was actually random. Also, the Dev Tox CSN showed a giant component phase transition as expected from an ER model. However, by definition, a random network should not show any kind of assortative behaviour: the assortativity of the Dev Tox CSN incontrovertibly demonstrates that it is not a random network, at least in the connection probability range explored. Moreover, its behaviour is far from that of a random network as the cut-off values approaches criticality. Thus, thresholding becomes intrinsically related to meaningfulness.
Once established the meaningfulness of the Dev Tox CSN, we characterised its inner communities using both molecular descriptors, as similar chemicals in a physical, biological or toxicological sense tend to exhibit similar properties62,63,64,65, and biological functions. The proposed CSN is thus easily interpretable by domain experts34,66 and could be profitably employed for drug repurposing and rational de novo design67, strategic assets to mitigate the well-known issues of drug discovery, such as huge costs and extremely time-consuming procedures68,69,70. In fact, thanks to PLATO target profiling, the Dev Tox archetypes outlined within the CSN could be related (with different reliability) to multiple biological activities.
This work also investigates the predictive power of the proposed CSN. The model was able to reach a reliable accuracy in Dev Tox prediction, with extremely high sensitivity. On the contrary, specificity remained substantially low. Two aspects deserve to be considered: (i) the informative content provided by structural similarity cannot reasonably provide a comprehensive description of toxicological patterns. It is easy to find examples of chemicals characterised by high structural similarity which show opposite toxicological behaviours, e.g., Dydrogesterone and Progesterone, non-toxic and toxic, respectively, differ by only a double bond56. (ii) While toxicants are expected to have common characteristics, the wide spectrum of chemicals which are non-toxic with respect to this specific endpoint include very heterogeneous chemotypes, which in principle can share few or even no structural similarities. Thus, for a classification model, the correct detection of non-toxic chemicals is extremely challenging, not to mention the fact that this class is poorly represented in the available databases. It is worth mentioning that this is not an unexpected behaviour, it has been already observed in literature, although by studies based on different descriptions31,32,33,71,72,73.
As a final remark, it is worth noting how complex network software suites and methodologies can manage systems with millions of nodes and therefore, thanks to its generality, the proposed approach can be straightforwardly adopted for broader chemical spaces, not necessarily limited to a single endpoint, and devoted to several applications. Our findings suggest that the characterization of the CSN could support in silico assessment of chemicals, specifically the so-called New Approach Methodologies (NAMs). The combined use of features derived from the CSN along with physicochemical descriptors and fingerprints could in principle enhance existing models.
Methods
Data curation
The chemicals were downloaded in SMILES format, with the associated binary Dev Tox label, from the freely accessible CAESAR and P&G databases. All SMILES data were cleaned of stereoisomeric assignments, were canonised and then the two databases were cross merged. 8 matches with opposite labels, derived mainly from P&G dataset, were discarded from the analysis; finally, 684 toxic molecules were collected. The selected chemicals were described by 2D molecular descriptors obtained from RDKit, and autocorrelators obtained from Mordred. Descriptors with a variability lower than 10% were removed from the analysis, thus resulting in a total of 774 descriptors, see the Table S3 of the Supporting Information for a comprehensive list.
Network analyses
The SMSD computes the largest common subgraph between two chemicals, where the molecular graph is a natural representation of a molecule based on its bonds and its atoms, except for hydrogen atoms, which are treated as implicit. The SMSD Tanimoto measure is therefore the ratio of the size of the common subgraph between two molecule and the size of the union of in common and not in common subgraphs. We used the implementation based on current CDK available at https://mvnrepository.com/artifact/gov.nih.ncats/smsd-core.
The CSN was compared with a uniform ER model, specifically an ER model G(N,E) whose E edges are uniformly sampled among the N(N-1)/2 possible connections. The advantage of such model is to provide a graph with the same number of connections as the thresholded CSN.
Comparisons were carried out by considering three centrality metrics, accounting for three different perspectives: a local, a global and a dynamic one.
-
Degree ({d}_{i}) of node i (local metric). ({d}_{i}={sum }_{j=1}^{N}{a}_{ij}) with ({a}_{ij}) representing the elements of the adjacency matrix of the considered network having N nodes. The degree takes into account only the connections of a node, in this sense it is a local centrality metric.
-
Eigenvector centrality ({e}_{i}) of node i (global metric). ({e}_{i}=left(frac{1}{lambda }right){sum }_{j=1}^{N}{a}_{ij}{e}_{j}) so that ({e}_{i}) satisfies an eigenvalue equation.
-
Betweenness ({b}_{i}) of node i (dynamic metric). ({b}_{i}={sum }_{sne ine t}^{N}frac{{pleft(iright)}_{st}}{{p}_{st}}), which measures the ratio between the number of paths p connecting a generic pair of nodes (s,t) passing through the node i and all the paths connecting them. Thus, this centrality metric evaluates the dynamical information flow within the network.
Moreover, to emphasise the difference between the defined CSN and a random network, assortativity was used.
-
Assortativity measures the preference of nodes to be connected to other nodes according to a similarity criterion, e.g., degree. For example, in assortative networks, high degree nodes tend to connect to nodes with high degree. On the other hand, if they prefer low-degree nodes, the network is anti-assortative. Random networks tend to have null assortativity as a consequence of connections’ randomness.
Studying these metrics allowed us to reveal the giant component phase transition. At criticality, community detection was performed by means of the Louvain algorithm74. All network analyses were carried out with a Python 3.8.13 distribution and the NetworkX 3.1 package.
Statistical significance
We evaluated the differences between the distributions of several molecular descriptors within the communities that arose at criticality. To this aim, to discard any a priori hypothesis about the descriptor distributions, the non-parametric Mood’s test for medians was performed. Three tests were carried out for each descriptor to ensure that a significant difference had been found among all the three communities. The chosen significance level was 0.01 but a Bonferroni correction was adopted to avoid the multiple comparison bias.
The PLATO platform for target profiling
The predictive web platform PLATO was used to predict relevant therapeutic drug targets of small molecules. PLATO matches query molecules with the most similar molecules in its database of experimental activity values based on Tanimoto similarity calculated on 13 different fingerprints. The predicted targets with their referenced organism are experimentally linked to the similar molecules identified by the algorithm. For each prediction, a score is calculated by summing the Tanimoto coefficients of each fingerprint. This implies that an exact match corresponds to a score equal to 13. Query reports can be easily provided in json format upon programmatic POST requests. PLATO is freely available at https://prometheus.farmacia.uniba.it/plato/.
Data availability
Data used in this work are publicly available. The entire list of Dev Tox chemicals (in SMILES format) herein analysed is provided in the Table S1 of the Supporting Information.
References
-
Maggiora, G. M. & Bajorath, J. Chemical space networks: A powerful new paradigm for the description of chemical space. J. Comput. Aided Mol. Des. 28, 795–802 (2014).
Google Scholar
-
Vogt, M., Stumpfe, D., Maggiora, G. M. & Bajorath, J. Lessons learned from the design of chemical space networks and opportunities for new applications. J. Comput. Aided Mol. Des. 30, 191–208 (2016).
Google Scholar
-
Scalfani, V. F., Patel, V. D. & Fernandez, A. M. Visualizing chemical space networks with RDKit and NetworkX. J. Cheminform. 14, 87 (2022).
Google Scholar
-
Recanatini, M. & Cabrelle, C. Drug research meets network science: Where are we?. J. Med. Chem. 63, 8653–8666 (2020).
Google Scholar
-
Kunimoto, R. & Bajorath, J. Combining similarity searching and network analysis for the identification of active compounds. ACS Omega 3, 3768–3777 (2018).
Google Scholar
-
Kunimoto, R. & Bajorath, J. Exploring sets of molecules from patents and relationships to other active compounds in chemical space networks. J. Comput. Aided Mol. Des. 31, 779–788 (2017).
Google Scholar
-
Medina-Franco, J. L., Sánchez-Cruz, N., López-López, E. & Díaz-Eufracio, B. I. Progress on open chemoinformatic tools for expanding and exploring the chemical space. J. Comput. Aided Mol. Des. 36, 341–354 (2022).
Google Scholar
-
Villoutreix, B. O., Labbé, C. M., Lagorce, D., Laconde, G. & Sperandio, O. A leap into the chemical space of protein-protein interaction inhibitors. Curr. Pharm. Des. 18, 4648–4667 (2012).
Google Scholar
-
Kothiyal, M., Kumar, S. & Sukumar, N. Investigation of chemical space networks using graph measures and random matrix theory. J. Math. Chem. 60, 891–914 (2022).
Google Scholar
-
Barabási, A.-L. & Bonabeau, E. Scale-free networks. Sci. Am. 288, 60–69 (2003).
Google Scholar
-
Watts, D. J. & Strogatz, S. H. Collective dynamics of ‘small-world’ networks. Nature 393, 440–442 (1998).
Google Scholar
-
Janson, S., Luczak, T. & Rucinski, A. Random Graphs (Wiley, 2011).
Google Scholar
-
Zwierzyna, M., Vogt, M., Maggiora, G. M. & Bajorath, J. Design and characterization of chemical space networks for different compound data sets. J. Comput. Aided Mol. Des. 29, 113–125 (2015).
Google Scholar
-
Zhang, B., Vogt, M., Maggiora, G. M. & Bajorath, J. Design of chemical space networks using a Tanimoto similarity variant based upon maximum common substructures. J. Comput. Aided Mol. Des. 29, 937–950 (2015).
Google Scholar
-
Gurunathan, S., Yogalakshmi, T. & Balasubramanian, K. Topological characterization of statistically clustered networks for molecular similarity analysis. J. Math. Chem. 61, 859–876 (2023).
Google Scholar
-
Dunn, T. B. et al. Diversity and chemical library networks of large data sets. J. Chem. Inf. Model. 62, 2186–2201 (2022).
Google Scholar
-
Saldívar-González, F. I. & Medina-Franco, J. L. Approaches for enhancing the analysis of chemical space for drug discovery. Expert Opin. Drug Discov. 17, 789–798 (2022).
Google Scholar
-
Boezio, B., Audouze, K., Ducrot, P. & Taboureau, O. Network-based approaches in pharmacology. Mol. Inform. https://doi.org/10.1002/minf.201700048 (2017).
Google Scholar
-
Liu, H. Comparison between Merrifield-Simmons index and some vertex-degree-based topological indices. Comp. Appl. Math. 42, 89 (2023).
Google Scholar
-
Wu, M., Vogt, M., Maggiora, G. M. & Bajorath, J. Design of chemical space networks on the basis of Tversky similarity. J. Comput. Aided Mol. Des. 30, 1–12 (2016).
Google Scholar
-
Dorogovtsev, S. N., Goltsev, A. V. & Mendes, J. F. F. Critical phenomena in complex networks. Rev. Mod. Phys. 80, 1275–1335 (2008).
Google Scholar
-
Radin, C. & Sadun, L. Phase transitions in a complex network. J. Phys. A: Math. Theor. 46, 305002 (2013).
Google Scholar
-
Bollobás, B., Janson, S. & Riordan, O. The phase transition in inhomogeneous random graphs. Random Struct. Algorithms 31, 3–122 (2007).
Google Scholar
-
Newman, M. E. J. & Watts, D. J. Renormalization group analysis of the small-world network model. Phys. Lett. A 263, 341–346 (1999).
Google Scholar
-
Fox Keller, E. Revisiting, “scale-free” networks. BioEssays 27, 1060–1068 (2005).
Google Scholar
-
Amoroso, N. et al. Potential energy of complex networks: A quantum mechanical perspective. Sci. Rep. 10, 18387 (2020).
Google Scholar
-
Gupta, P. K. Developmental toxicology. In Problem Solving Questions in Toxicology: A Study Guide for the Board and Other Examinations (ed. Gupta, P. K.) 137–143 (Springer International Publishing, 2020). https://doi.org/10.1007/978-3-030-50409-0_10.
Google Scholar
-
Merlot, C. Computational toxicology—A tool for early safety evaluation. Drug Discov. Today 15, 16–22 (2010).
Google Scholar
-
Guengerich, F. P. & MacDonald, J. S. Applying mechanisms of chemical toxicity to predict drug safety. Chem. Res. Toxicol. 20, 344–369 (2007).
Google Scholar
-
Cassano, A. et al. CAESAR models for developmental toxicity. Chem. Central J. 4, S4 (2010).
Google Scholar
-
Gunturi, S. B. & Ramamurthi, N. A novel approach to generate robust classification models to predict developmental toxicity from imbalanced datasets. SAR QSAR Environ. Res. 25, 711–727 (2014).
Google Scholar
-
Zhang, H., Mao, J., Qi, H.-Z. & Ding, L. In silico prediction of drug-induced developmental toxicity by using machine learning approaches. Mol. Divers. 24, 1281–1290 (2020).
Google Scholar
-
Togo, M. V. et al. TIRESIA: An explainable artificial intelligence platform for predicting developmental toxicity. J. Chem. Inform. Model. https://doi.org/10.1021/acs.jcim.2c01126 (2022).
Google Scholar
-
Jiménez-Luna, J., Grisoni, F. & Schneider, G. Drug discovery with explainable artificial intelligence. Nat. Mach. Intell. 2, 573–584 (2020).
Google Scholar
-
Iswarya, B. & Manimekalai, K. Drug discovery with XAI using deep learning. In Principles and Methods of Explainable Artificial Intelligence in Healthcare (eds de Albuquerque, V. H. C. et al.) 131–149 (IGI Global, 2022). https://doi.org/10.4018/978-1-6684-3791-9.ch006.
Google Scholar
-
Kikutsuji, T. et al. Explaining reaction coordinates of alanine dipeptide isomerization obtained from deep neural networks using Explainable Artificial Intelligence (XAI). J. Chem. Phys. 156, 154108 (2022).
Google Scholar
-
Cilli, R. et al. Explainable artificial intelligence (XAI) detects wildfire occurrence in the Mediterranean countries of Southern Europe. Sci. Rep. 12, 16349 (2022).
Google Scholar
-
Wu, S. et al. Framework for identifying chemicals with structural features associated with the potential to act as developmental or reproductive toxicants. Chem. Res. Toxicol. 26, 1840–1861 (2013).
Google Scholar
-
Hewitt, M., Ellison, C. M., Enoch, S. J., Madden, J. C. & Cronin, M. T. D. Integrating (Q)SAR models, expert systems and read-across approaches for the prediction of developmental toxicity. Reprod. Toxicol. 30, 147–160 (2010).
Google Scholar
-
Freyer, A. M. Drugs in pregnancy and lactation 8th edition: A reference guide to fetal and neonatal risk. Obstet. Med. 2, 89 (2009).
Google Scholar
-
Rahman, S. A., Bashton, M., Holliday, G. L., Schrader, R. & Thornton, J. M. Small Molecule Subgraph Detector (SMSD) toolkit. J. Cheminform. 1, 12 (2009).
Google Scholar
-
Bajusz, D., Rácz, A. & Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations?. J. Cheminform. 7, 20 (2015).
Google Scholar
-
Boccaletti, S., Latora, V., Moreno, Y., Chavez, M. & Hwang, D.-U. Complex networks: Structure and dynamics. Phys. Rep. 424, 175–308 (2006).
Google Scholar
-
Combining complex networks and data mining: Why and how – ScienceDirect | https://www.sciencedirect.com/.
-
Prediction of Physicochemical Parameters by Atomic Contributions | Journal of Chemical Information and Modeling. https://pubs.acs.org/.
-
Bickerton, G. R., Paolini, G. V., Besnard, J., Muresan, S. & Hopkins, A. L. Quantifying the chemical beauty of drugs. Nat. Chem. 4, 90–98 (2012).
Google Scholar
-
Consonni, V. & Todeschini, R. Molecular descriptors. In Challenges and Advances in Computational Chemistry and Physics (eds Puzyn, T. et al.) 29–102 (Springer, 2009).
-
Moreau, G. & Broto, P. Autocorrelation of molecular structures, application to SAR studies. Nouv. J. Chim. 4, 757–764 (1980).
-
Nicolotti, O. & Carotti, A. QSAR and QSPR studies of a highly structured physicochemical domain. J. Chem. Inf. Model. 46, 264–276 (2006).
Google Scholar
-
Sliwoski, G., Mendenhall, J. & Meiler, J. Autocorrelation descriptor improvements for QSAR: 2DA_Sign and 3DA_Sign. J. Comput. Aided Mol. Des. 30, 209–217 (2016).
Google Scholar
-
Ciriaco, F., Gambacorta, N., Trisciuzzi, D. & Nicolotti, O. PLATO: A predictive drug discovery web platform for efficient target fishing and bioactivity profiling of small molecules. Int. J. Mol. Sci. 23, 5245 (2022).
Google Scholar
-
The Chemical Space Project | Accounts of Chemical Research. https://doi.org/10.1021/ar500432k.
-
Reymond, J.-L., van Deursen, R., Blum, L. C. & Ruddigkeit, L. Chemical space as a source for new drugs. Med. Chem. Commun. 1, 30–38 (2010).
Google Scholar
-
Kirkpatrick, P. & Ellis, C. Chemical space. Nature 432, 823–823 (2004).
Google Scholar
-
Lipinski, C. & Hopkins, A. Navigating chemical space for biology and medicine. Nature 432, 855–861 (2004).
Google Scholar
-
Cruz-Monteagudo, M. et al. Activity cliffs in drug discovery: Dr Jekyll or Mr Hyde?. Drug Discov. Today 19, 1069–1080 (2014).
Google Scholar
-
Gromski, P. S., Henson, A. B., Granda, J. M. & Cronin, L. How to explore chemical space using algorithms and automation. Nat. Rev. Chem. 3, 119–128 (2019).
Google Scholar
-
Coley, C. W. Defining and exploring chemical spaces. TRECHEM 3, 133–145 (2021).
-
Eckert, H. & Bajorath, J. Molecular similarity analysis in virtual screening: Foundations, limitations and novel approaches. Drug Discov. Today 12, 225–233 (2007).
Google Scholar
-
Scior, T. et al. How to recognize and workaround pitfalls in QSAR studies: A critical review. Curr. Med. Chem. 16, 4297–4313 (2009).
Google Scholar
-
Sahigara, F. et al. Comparison of different approaches to define the applicability domain of QSAR models. Molecules 17, 4791–4810 (2012).
Google Scholar
-
Hartenfeller, M. & Schneider, G. De novo drug design. In Chemoinformatics and Computational Chemical Biology (ed. Bajorath, J.) 299–323 (Humana Press, 2011). https://doi.org/10.1007/978-1-60761-839-3_12.
Google Scholar
-
Öztürk, H., Özgür, A., Schwaller, P., Laino, T. & Ozkirimli, E. Exploring chemical space using natural language processing methodologies for drug discovery. Drug Discov. Today 25, 689–705 (2020).
Google Scholar
-
Medina-Franco, J. L., Martinez-Mayorga, K., Giulianotti, M. A., Houghten, R. A. & Pinilla, C. Visualization of the chemical space in drug discovery. Curr. Comput. Aided Drug Des. 4, 322–333 (2008).
Google Scholar
-
Dobson, C. M. Chemical space and biology. Nature 432, 824–828 (2004).
Google Scholar
-
Askr, H. et al. Deep learning in drug discovery: An integrative review and future challenges. Artif. Intell. Rev. 56, 5975–6037 (2023).
Google Scholar
-
Alberga, D. et al. De Novo drug design of targeted chemical libraries based on artificial intelligence and pair-based multiobjective optimization. J. Chem. Inf. Model. 60, 4582–4593 (2020).
Google Scholar
-
Choudhury, C., Arul Murugan, N. & Priyakumar, U. D. Structure-based drug repurposing: Traditional and advanced AI/ML-aided methods. Drug Discov. Today 27, 1847–1861 (2022).
Google Scholar
-
Medina-Franco, J. L., Martinez-Mayorga, K. & Meurice, N. Balancing novelty with confined chemical space in modern drug discovery. Expert Opin. Drug Discov. 9, 151–165 (2014).
Google Scholar
-
Strittmatter, S. M. Overcoming drug development bottlenecks with repurposing: Old drugs learn new tricks. Nat. Med. 20, 590–591 (2014).
Google Scholar
-
Marzo, M. et al. Integrating in silico models to enhance predictivity for developmental toxicity. Toxicology 370, 127–137 (2016).
Google Scholar
-
Marzo, M., Roncaglioni, A., Kulkarni, S., Barton-Maclaren, T. S. & Benfenati, E. In silico models for developmental toxicity. In In Silico Methods for Predicting Drug Toxicity (ed. Benfenati, E.) 217–240 (Springer, 2022). https://doi.org/10.1007/978-1-0716-1960-5_10.
Google Scholar
-
Zhang, H. et al. Development of novel in silico model for developmental toxicity assessment by using naïve Bayes classifier method. Reprod. Toxicol. 71, 8–15 (2017).
Google Scholar
-
Blondel, V. D., Guillaume, J.-L., Lambiotte, R. & Lefebvre, E. Fast unfolding of communities in large networks. J. Stat. Mech. 2008, P10008 (2008).
Google Scholar
Acknowledgements
The authors are thankful to the following: Programma Operativo Nazionale Ricerca e Innovazione 2014-2020 (CCI 2014IT16M2OP005), Fondo Sociale Europeo, Azione I.1 “Dottorati Innovativi con caratterizzazione Industriale” the Comune di Bovino and PIANO STRALCIO <> 2015-2017, Comune di Bovino (Foggia, Italy) and Dott. Renato Lombardi, Director of “Struttura Complessa di Farmacia, IRCCS, Casa Sollievo della Sofferenza”, (Foggia, Italy) (Code: DOT19C9KX4); Horizon Europe Seeds “L’intelligenza artificiale a tutela della salute in età pediatrica. Implementazione di una piattaforma digitale per il design di farmaci pediatrici sicuri”, Università degli Studi di Bari (Bari, Italy) (CUP: H99J21017390006).
Author information
Authors and Affiliations
Contributions
N.A. conceived the study, carried out the analyses and wrote the main manuscript. F.M. and M.V.T. carried out the analyses and wrote the main manuscript. O.N. supervised the analysis and wrote the main manuscript. F.C. carried out the analyses. All the authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Supplementary Figure S1.
Supplementary Table S1.
Supplementary Table S2.
Supplementary Table S3.
Supplementary Legends.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Reprints and Permissions
About this article
Cite this article
Amoroso, N., Gambacorta, N., Mastrolorito, F. et al. Making sense of chemical space network shows signs of criticality.
Sci Rep 13, 21335 (2023). https://doi.org/10.1038/s41598-023-48107-3
-
Received: 13 September 2023
-
Accepted: 22 November 2023
-
Published: 04 December 2023
-
DOI: https://doi.org/10.1038/s41598-023-48107-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.