Abstract
Urban lakes provide multiple benefits to society while influencing life quality. Moreover, lakes and their microbiomes are sentinels of anthropogenic impact and can be used for natural resource management and planning. Here, we release original metagenomic data from several well-characterized and anthropogenically impacted eutrophic lakes in the vicinity of Stockholm (Sweden). Our goal was to collect representative microbial community samples and use shotgun sequencing to provide a broad view on microbial diversity of productive urban lakes. Our dataset has an emphasis on Lake Mälaren as a major drinking water reservoir under anthropogenic impact. This dataset includes short-read sequence data and metagenome assemblies from each of 17 samples collected from eutrophic lakes near the greater Stockholm area. We used genome-resolved metagenomics and obtained 2378 metagenome assembled genomes that de-replicated into 514 species representative genomes. This dataset adds new datapoints to previously sequenced lakes and it includes the first sequenced set of metagenomes from Lake Mälaren. Our dataset serves as a baseline for future monitoring of drinking water reservoirs and urban lakes.
Background & Summary
Healthy lakes and shorelines provide multiple societal benefits and contribute positively to our quality of life and livelihoods. Lakes can be used as sources of drinking water for surrounding urban areas and can also supply water for industry and agricultural irrigation. Lakes also offer ample opportunities for recreation and tourism. However, urbanization of surrounding areas, causing eutrophication and other types of anthropogenic impacts, can pose major threats to the sustainable use of these natural ecosystems. In this way, lakes are not only valuable resources, but also sentinels of anthropogenic impacts and environmental change, as their microbiomes are highly sensitive to perturbations, and respond rapidly and predictably to changing environmental conditions1,2,3,4. In depth and high-quality records of the current state of lake microbiomes can thus be used as a baseline to assess change and anthropogenic impacts on lake water quality. However, we face a paucity of such metagenomic data that could provide us with more deep and insightful information about microbial diversity and the genome-encoded functional traits of such communities. Here, we release metagenomic data (Table 1 and Table S1) and metagenome-assembled genomes (MAGs)5 from several urban and anthropogenically impacted Swedish lakes. Most of these lakes have previously been studied and characterized in terms of limnological features and water chemistry, but information on their microbial communities is scarce.
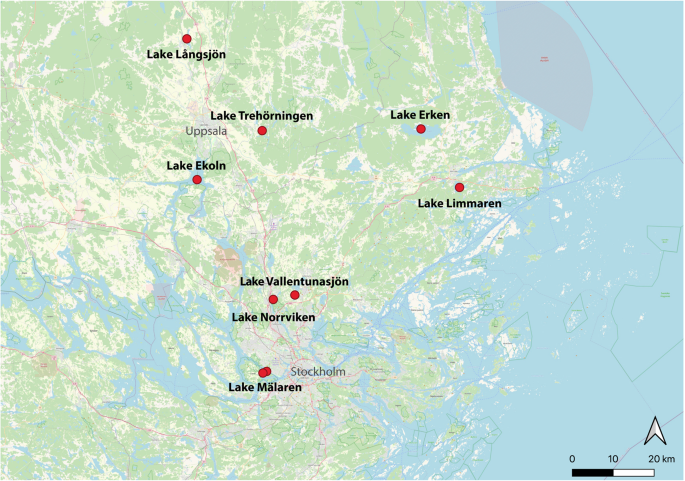
Lake Mälaren is the third largest lake in Sweden, and according to the Mälaren Water Protection Association (Mälarens vattenvårdsförbund), it serves as the main drinking water supply to approximately 2 million residents in Sweden. The lake receives high nutrient loads from surrounding agricultural areas and has a history of recurrent cyanobacterial blooms6. Moreover, the eastern part of the lake drains into the Baltic Sea, transporting nutrient rich waters into the vulnerable coastal zones7. Despite its significance, Lake Mälaren is severely understudied and the microbial community composition in the lake has only been superficially characterized. This comprehensive metagenomic dataset is thus the first detailed insight into the bacterial dynamics of Lake Mälaren, and was obtained during the 2021 summer season. For comparison, we sampled two productive lakes (Trehörningen and Långsjön; Fig. 1) in the vicinity of Uppsala to contrast and compare variation in microbial communities both within and between similar lakes from the same region of Sweden.

Map of sampling locations. Note that Mälaren was sampled at two nearby but different locations (named Mälaren_B and Mälaren_D in supplementary data files). For coordinates and metadata, see Table S1.
Additionally, we sequenced and assembled metagenomes from five previously sampled eutrophic lakes in the urban Stockholm-Uppsala region: lakes Ekoln, Erken, Limmaren, Vallentunasjön, and Norrviken. These samples, collected in 2002, have previously been characterized for their bacterial composition using less comprehensive and now outdated methods (i.e., clone libraries and sanger sequencing, and terminal-restriction fragment length polymorphism, T-RFLP)8,9. These five highly eutrophic lakes have a long history of seasonal cyanobacterial blooms in the summer8 and feature pronounced seasonal dynamics within the bacterioplankton communities9,10. In brief, Lake Ekoln is a subbasin in the northern part of Lake Mälaren. It stratifies in summer and receives high nutrient inputs from the city of Uppsala and the surrounding agricultural areas. Lake Erken, located east of Uppsala, is a eutrophic lake that is thermally stratified during the summer and has been extensively studied for several decades11, also with regards to microbial community composition12,13,14. Lake Erken also serves as a backup drinking water reservoir for the nearby city of Norrtälje. Lake Limmaren, located 70 km north of Stockholm, receives high nutrient loads from sediments and has a long history of dense blooms of Microcystis, Anabaena, and Aphanizomenon8. Historical accumulation of nutrients from urban settlements also plays a significant role in the state of the hypereutrophic lake Vallentunasjön located in a suburban area north of Stockholm10. The lake has undergone major restoration efforts, but still suffers from eutrophication with frequent cyanobacterial blooms. Lastly, lake Norrviken in Stockholm has received high historical loads of domestic and industrial sewage in the past and is also subjected to intense cyanobacterial blooms15.
Our broader ambition was to collect and sequence data that could be used to provide a comprehensive view of microbial communities in urban lakes of the greater Stockholm area, with special emphasis on Lake Mälaren (Table S1). Such data could also be used to identify microbial health hazards (such as pathogens), serving as a baseline for future monitoring efforts based on microbiomes as sentinels of environmental health. Finally, this dataset could be used to generate novel hypotheses on linkages between lake microbiomes and human activities in the watershed. We thus release 17 shotgun metagenomes (Table S2) and their corresponding single-sample assemblies. In addition, we performed genome-resolved metagenomics and obtained 2378 MAGs (>30% completeness and <10% contamination) (Fig. 2 and Table S3). We then clustered MAGs from across all samples together based on 95% average nucleotide identity (ANI) and obtained 514 species representative genomes (SRGs; >50% completeness and <6% contamination) (Fig. 2). We also provide an overview of the number of SRGs specific to and shared between the different sampled lakes (Fig. 3) and relative abundance patterns of different classes of Bacteria represented by SRGs across the lake metagenomes (Fig. 4 and Table S4).

Quality of the MAGs. Completeness (A) and contamination (B) across all the 2378 metagenome-assembled genomes (MAGs, in grey). Highlighted in black, the 514 species representative genomes (SRGs). Correlation between completeness and contamination for all 514 SRGs, colored by phyla (C). The legend table indicates the number of MAGs and SRGs classified as high-quality (>90% completeness and <5% contamination), medium-quality (≥50% completeness and <10% contamination), and low-quality (<50% completeness and <10% contamination), following the MIMAG standards for MAG quality completeness and contamination cutoffs35.

Intersection plot of shared species representative genomes (SRGs) across samples. Intersection size shows the number of SRGs present in each sample. The sum of all intersection sizes equals the 514 SRGs in the dataset. For example, we find that 60 SRGs are exclusively present in Limmaren Sample 101 S75. Set size is the number of SRGs present in total per sample. Samples highlighted in blue are from Lake Mälaren.

Heatmap indicating the sum of genome copies per million reads per class for each bacterial class represented by the SRGs per sample. White indicates that reads from the given class were not detected in the sample. In the figure, the color yellow/khaki starts at 0.0001 genome copies per million reads. The heatmap is hierarchically clustered.
Methods
Sampling
Surface water samples from Swedish lakes were collected in both 20028 and 2021 (Fig. 1). Samples from lakes Ekoln, Erken, Limmaren, Norrviken, and Vallentunasjön were collected in August 2002 and their bacterial 16 S rRNA gene composition was previously superficially described using molecular cloning and sanger sequencing8. We retrieved one membrane filter (Supor, Gelman) from each of those lakes from a −80 °C freezer, where they have been stored since 2002. Samples from lakes Mälaren (2 locations in Stockholm: Drottningholm – D and Brostugan – B), Trehörningen (Uppsala), and Långsjön (Uppsala) were collected in July and August 2021 (Table 1). In these cases, surface water was collected from a wooden deck with a Limnos tube-sampler (Limnos, Poland) and 300 mL hand-filtered in duplicate onto 0.2 µm Sterivex filters (Millipore) that were subsequently frozen at −20 °C until DNA extraction. Environmental parameters (temperature, dissolved oxygen, and conductivity) were measured using a YSI sonde (Table S1).
DNA extractions
For all samples, DNA was extracted using the DNAeasy PowerWater kit (Qiagen) following the manufacturer’s instructions and DNA concentrations were measured using a Qubit dsDNA HS kit (Thermo Fisher Scientific Inc). However, for some of the samples the Qiagen kit did not yield high quality DNA. Additionally, the duplicate filters from 2021 were selected for extractions at Linneaus University using the FastDNA® SPIN Kit for soil (MP Biomedicals) with a modified cell lysis step to ensure extraction of cyanobacteria. First, 1467 µL of Sodium Phosphate Buffer (from kit), 183 µl MT buffer (from kit), and 16.5 µl of Proteinase K solution (MP Biomedicals) were added to the Sterivex filters. The filters were mixed, tightly capped, wrapped in parafilm and incubated overnight (~15 hours) in a rotating oven at 55 °C. This was done to ensure extraction of DNA from low biomass samples and to improve cell lysis. Following the overnight incubations, samples were extracted following the manufacturer’s protocol. All DNA concentrations were measured using the Qubit dsDNA HS kit (Thermo Fisher Scientific Inc). All samples that yielded DNA were sent for sequencing.
Library preparation and sequencing
Sequence libraries were prepared using SMARTer Thruplex library preparation (350 bp average fragment size) at the National Genomics Infrastructure (NGI) at the Science for Life Laboratory (SciLifeLab) in Stockholm. Sequencing was done on the Illumina NovaSeq 6000 platform using a S4 v1.5 flowcell in 300 cycle mode (2 × 150 bp). The Bcl to FastQ conversion was performed using bcl2fastq_v2.20.0.422 from the CASAVA software suite at NGI. Sequences were demultiplexed and quality control and raw data were retrieved on an HPC server hosted by the Swedish National Infrastructure for Computing (SNIC).
Analysis of raw sequence reads
Processing of raw sequence reads was performed using the metaWRAP pipeline16 (v1.3.2). Forward and reverse reads were first trimmed using the “read_qc” module with default settings and TrimGalore17 (v0.5.0). Final trimmed reads were assembled into metagenome assemblies using the “metaWRAP_assembly” module with MegaHit18 (v1.1.3). Short scaffolds (<1000 bp) were discarded by default before assessing assembly quality statistics using QUAST19 (v. 5.0.2) (Table S2). Assembly of contigs were subsequently binned into MAGs using the “metaWRAP_binning” module. Briefly, this module performs binning using three metagenomic binning tools: CONCOCT20 (v1.0), metaBAT221 (v2.12.1) and maxBIN222 (v2.2.6). Bins generated from these three tools were then consolidated and refined using the “metaWRAP_bin_refinement” module with cutoffs of above 30% for completeness and below 10% for contamination, resulting in MAGs (Table S3). The final bin set was assessed by CheckM23 (v1.1.3) for completeness, contamination, and other statistics. All bins with quality completeness >30% and contamination <10% were considered as MAGs (for detailed statistics see Fig. 2) and included in further analyses.
Taxonomic classification of MAGs was performed using GTDB-tk24 (v1.5.0) according to GTDB classification25 (data release version r202). Taxonomy of all 2378 MAGs can be found in Table S3. All MAGs were then dereplicated at the species-level using dRep26 (v3.0.0) with default settings, which resulted in 514 genomes that represent the species present across all of the lakes (SRGs; Fig. 3). In this pipeline, MAGs were first compared with a rapid primary algorithm MASH27 and then a secondary clustering algorithm ANIm was run based on an Average Nucleotide Identity (ANI) threshold of 95%, genome completeness of > = 50%, and contamination < = 5%. The most complete and least contaminated MAGs were selected as species representatives (Table S3). Finally, the “metaWRAP_quant_bin” module was used to estimate the relative abundance of SRGs across all samples. The “metaWRAP_quant_bins” module estimates the abundance of MAGs across the sampling using Salmon28 (v1.9.0) to index the metagenomic assembly and align reads from each sample to the assembly. Coverage tables were generated estimating the abundance of each contig in each sample in genome copies per million reads (Fig. 4 and Table S4).
To estimate the total number of reads mapped per metagenome, we mapped all 514 SRGs to all trimmed clean reads using Bowtie2 (v2.5.1)29. An index was created using the function bowtie2-build calling all 514 SRGs, and then mapped against all 17 metagenomes using default parameters. The resulting sam files were converted into bam files, and then used to count the number of mapped clean reads using SAMtools30 (v1.17). These results are reported in Table S1.
A map of the sampling locations was constructed in ArcGIS (v3.28 Firenze). Figures depicting SRG completeness and contamination were generated using ggplot2 (v3.3.5) in RStudio (v2022.02.3 + 492). The intersection graph of shared SRGs across samples was generated using the UpsetR package31 (v1.4.0). To obtain the total number of genome copies per million reads for every represented bacterial class (Fig. 4), we took the genome copies per million for each SRG (Table S4) and summed them per bacterial class. We then calculated the number of SRGs per class using the function percencat from the package plada (v0.1.0; https://github.com/alejandrorgijon/plada_package). The heatmap was generated using the R package ComplexHeatmap32 (v2.10.0) and hierarchical clustering was performed with default settings.
Data Records
All raw read sequence files and single-sample metagenome assemblies are available at the European Nucleotide Archive (ENA) under the BioProject accession PRJEB5481733. All 2378 MAGs have been deposited in a SciLifeLab Figshare data repository5: https://doi.org/10.17044/scilifelab.22270225.v3. The 514 SRGs have also been deposited under NCBI Bioproject PRJNA102139134. Statistics for raw reads, assemblies, refined high-quality bins, and dereplicated MAGs (SRGs) are provided in the supplementary tables, and include MAG and SRG IDs and taxonomy, MAG membership in SRGs, presence and relative abundance estimates of SRGs across samples, and genome information (Tables S1–S4).
Technical Validation
The quality of the raw reads was monitored and certified by the National Genomics Infrastructure (NGI) in Solna, Sweden according to accreditation by Swedac ISO/IEC 17025. The quality scale used is Sanger/phred33/Illumina 1.8+. Quality distribution showed Q30 aggregated percentage of bases to be higher than 89 for all metagenomes. PHRED score was 36 for all samples (Table S1). The quality of the MAGs that compose the SRGs was computed with CheckM23 (v1.1.3).
Code availability
No custom code was used in this project.
References
-
Williamson, C. E., Saros, J. E., Vincent, W. F. & Smol, J. P. Lakes and reservoirs as sentinels, integrators, and regulators of climate change. Limnology and Oceanography 54, 2273–2282, https://doi.org/10.4319/lo.2009.54.6_part_2.2273 (2009).
Google Scholar
-
Cavicchioli, R. et al. 2019. Scientists’ warning to humanity: microorganisms and climate change. Nature Reviews Microbiology 17, 569–586, https://doi.org/10.1038/s41579-019-0222-5 (2019).
Google Scholar
-
Linz, A. M. et al. Bacterial community composition and dynamics spanning five years in freshwater bog lakes. mSphere 2, e00169–17, https://doi.org/10.1128/mSphere.00169-17 (2017).
Google Scholar
-
Kraemer, S. A. et al. A large-scale assessment of lakes reveals a pervasive signal of land use on bacterial communities. The ISME Journal 14, 3011–3023, https://doi.org/10.1038/s41396-020-0733-0 (2020).
Google Scholar
-
Garcia, S. & Hampel, JJ. Metagenomic dataset from Swedish urban lakes, SciLifeLab Data Repository, https://doi.org/10.17044/scilifelab.22270225.v3 (2023).
-
Willén, E. Four decades of research on the Swedish large lakes Mälaren, Hjälmaren, Vättern and Vänern: the significance of monitoring and remedial measures for a sustainable society. AMBIO: A Journal of the Human Environment 30, 458–466, https://doi.org/10.1579/0044-7447-30.8.458 (2001).
Google Scholar
-
Darracq, A., Greffe, F., Hannerz, F., Destouni, G. & Cvetkovic, V. Nutrient transport scenarios in a changing Stockholm and Mälaren valley region, Sweden. Water Science and Technology 51, 31–38, https://doi.org/10.2166/wst.2005.0572 (2005).
Google Scholar
-
Eiler, A. & Bertilsson, S. Composition of freshwater bacterial communities associated with cyanobacterial blooms in four Swedish lakes. Environmental Microbiology 6, 1228–1243, https://doi.org/10.1111/j.1462-2920.2004.00657.x (2004).
Google Scholar
-
Bertilsson, S., Eiler, A., Nordqvist, A. & Jørgensen, N. O. G. Links between bacterial production, amino-acid utilization and community composition in productive lakes. The ISME Journal 1, 532–544, https://doi.org/10.1038/ismej.2007.64 (2007).
Google Scholar
-
Eiler, A. & Bertilsson, S. Flavobacteria blooms in four eutrophic lakes: linking population dynamics of freshwater bacterioplankton to resource availability. Applied and Environmental Microbiology 73, 3511–3518, https://doi.org/10.1128/AEM.02534-06 (2007).
Google Scholar
-
Pettersson, K., Grust, K., Weyhenmeyer, G. & Blenckner, T. Seasonality of chlorophyll and nutrients in Lake Erken–effects of weather conditions. Hydrobiologia 506, 75–81, https://doi.org/10.1023/B:HYDR.0000008582.61851.76 (2003).
Google Scholar
-
Eiler, A., Heinrich, F. & Bertilsson, S. Coherent dynamics and association networks among lake bacterioplankton taxa. The ISME Journal 6, 330–342, https://doi.org/10.1038/ismej.2011.113 (2012).
Google Scholar
-
Mondav, R. et al. Streamlined and abundant bacterioplankton thrive in functional cohorts. mSystems 5, e00316–20, https://doi.org/10.1128/mSystems.00316-20 (2020).
Google Scholar
-
Buck, M. et al. Comprehensive dataset of shotgun metagenomes from oxygen stratified freshwater lakes and ponds. Scientific Data 8, 131, https://doi.org/10.1038/s41597-021-00910-1 (2021).
Google Scholar
-
Routh, J., Choudhary, P., Meyers, P. A. & Kumar, B. A sediment record of recent nutrient loading and trophic state change in Lake Norrviken, Sweden. Journal of Paleolimnology 42, 325–341, https://doi.org/10.1007/s10933-008-9279-2 (2009).
Google Scholar
-
Uritskiy, G. V., DiRuggiero, J. & Taylor, J. MetaWRAP—a flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 6, 1–13, https://doi.org/10.1186/s40168-018-0541-1 (2018).
Google Scholar
-
Krueger, F. et al. FelixKrueger/TrimGalore: v0.6.10 – add default decompression path (0.6.10). Zenodo. https://doi.org/10.5281/zenodo.7598955 (2023).
-
Li, D., Liu, C. M., Luo, R., Sadakane, K. & Lam, T. W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676, https://doi.org/10.1093/bioinformatics/btv033 (2015).
Google Scholar
-
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075, https://doi.org/10.1093/bioinformatics/btt086 (2013).
Google Scholar
-
Alneberg, J. et al. Binning metagenomic contigs by coverage and composition. Nature Methods 11, 1144–1146, https://doi.org/10.1038/nmeth.3103 (2014).
Google Scholar
-
Kang, D. D. et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ Preprints 7, e7359, https://doi.org/10.7287/peerj.preprints.27522v1 (2019).
Google Scholar
-
Wu, Y. W., Simmons, B. A. & Singer, S. W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607, https://doi.org/10.1093/bioinformatics/btv638 (2016).
Google Scholar
-
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P. & Tyson, G. W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Research 25, 1043–1055, https://doi.org/10.1101/gr.186072.114 (2015).
Google Scholar
-
Chaumeil, P. A., Mussig, A. J., Hugenholtz, P. & Parks, D. H. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927, https://doi.org/10.1093/bioinformatics/btz848 (2020).
Google Scholar
-
Parks, D. H. et al. A complete domain-to-species taxonomy for Bacteria and Archaea. Nature Biotechnology 38, 1079–1086, https://doi.org/10.1038/s41587-020-0501-8 (2020).
Google Scholar
-
Olm, M. R., Brown, C. T., Brooks, B. & Banfield, J. F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. The ISME Journal 11, 2864–2868, https://doi.org/10.1038/ismej.2017.126 (2017).
Google Scholar
-
Ondov, B. D. et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biology 17, 1–14, https://doi.org/10.1186/s13059-016-0997-x (2016).
Google Scholar
-
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A. & Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nature Methods 14, 417–419, https://doi.org/10.1038/nmeth.4197 (2017).
Google Scholar
-
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359, https://doi.org/10.1038/nmeth.1923 (2012).
Google Scholar
-
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience 10, p.giab008, https://doi.org/10.1093/gigascience/giab008 (2021).
Google Scholar
-
Conway, J. R., Lex, A. & Gehlenborg, N. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33, 2938–2940, https://doi.org/10.1093/bioinformatics/btx364 (2017).
Google Scholar
-
Gu, Z., Eils, R. & Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849, https://doi.org/10.1093/bioinformatics/btw313 (2016).
Google Scholar
-
ENA European Nucleotide Archive, https://identifiers.org/ena.embl:PRJEB54817 (2022).
-
NCBI BioProject https://identifiers.org/ncbi/bioproject:PRJNA1021391 (2023).
-
Bowers, R. et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nature Biotechnology 35, 725–731, https://doi.org/10.1038/nbt.3893 (2017).
Google Scholar
Acknowledgements
This work was funded by SciLifeLab, the Albert and Maria Bergstrom Foundation, and the Wenner-Gren Foundation (project UPD2021-0051 and UPD2020-0040). We thank the SciLifeLab Research Community Program “Aquatic Microbiome Research Initiative” (AMRI) for funding the research exchange and travel to Linnaeus University. We thank Hanna Farnelid, Laura Bas Conn, and Anabella Aguilera at Linnaeus University for protocol and support with MP Biomedical DNA extractions. The authors acknowledge support from the National Genomics Infrastructure (NGI) in Stockholm funded by Science for Life Laboratory, the Knut and Alice Wallenberg Foundation and the Swedish Research Council (VR). Additionally, sequencing, computational analyses, and data handling were enabled by resources provided by the Swedish National Infrastructure for Computing (SNIC) at UPPMAX under projects SNIC 2022/5-392, SNIC 2022/5-137 and SNIC 2022/6-77, partially funded by the Swedish Research Council (VR) through the grant agreement no. 2018-05973.
Funding
Open access funding provided by Stockholm University.
Author information
Authors and Affiliations
Contributions
S.L.G. and J.J.H. conceived the project idea and secured funding. J.J.H., S.L.G., A.R.-G. and S.B. sampled lakes in 2021 and S.B. provided samples from 2002. J.J.H., J.E.D. and A.R.-G. performed computational analyses. A.R.-G. submitted all metagenomic and genomic data to public repositories. J.J.H., A.R.-G. and S.L.G. drafted the manuscript. All the authors contributed significant inputs to the final manuscript. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Table S1
Table S2
Table S3
Table S4
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
Reprints and Permissions
About this article
Cite this article
Rodríguez-Gijón, A., Hampel, J.J., Dharamshi, J. et al. Shotgun metagenomes from productive lakes in an urban region of Sweden.
Sci Data 10, 810 (2023). https://doi.org/10.1038/s41597-023-02722-x
-
Received: 04 April 2023
-
Accepted: 06 November 2023
-
Published: 17 November 2023
-
DOI: https://doi.org/10.1038/s41597-023-02722-x